Next generation sequencing can produce anywhere from hundreds of thousands to tens of millions short nucleotide sequences for a single sample. For any given base within an individual sequence there can also be a quality score associated with the confidence of that base call from the sequencer. The process of alignment is used to map all of these reads to a reference sequence, providing information with regards to the start and stop positions of each read within the reference sequence as well as a quality metric for the mapping. This document will provide information about the available aligners within Partek Flow as well as illustrate how to perform alignment against a reference sequence. The result of alignment will be an Aligned reads data node that contains the BAM files generated from the alignment.
The user should be familiar with:
Alignment tools appear in the context-sensitive menu on the right of the screen (Figure 1) when click on any data node containing FASTQ files. Examples include Unaligned reads, Trimmed reads, and Subsampled reads data nodes.
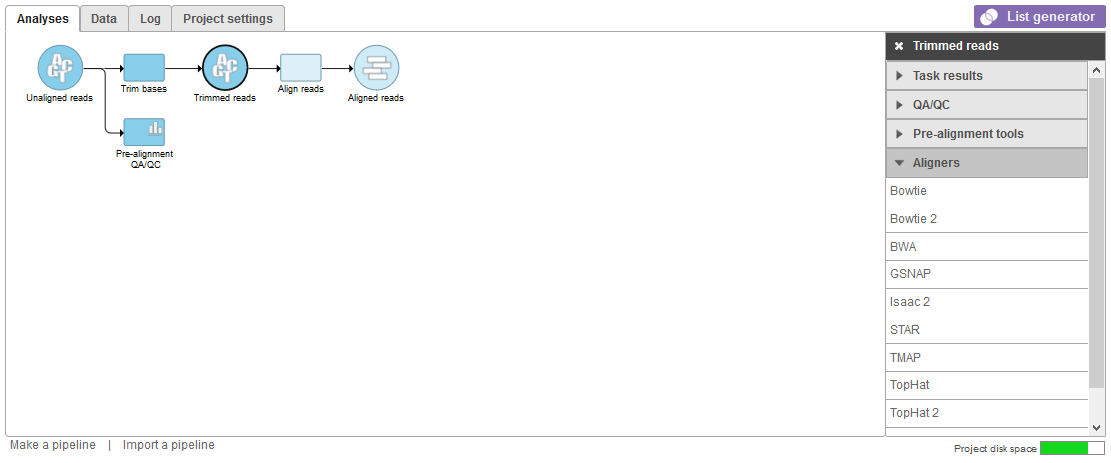
Figure 1. Showing Aligners from a trimmed reads node
Partek® Flow® provides numerous publicly available tools for the alignment process to meet the needs of your specific sequencing experiment. The information below provides a synopsis of each aligner as well as the current version. Please refer to the aligner links and references section for further information on each aligner.
Bowtie1 (Version 1.0.0) - Uses a Burrows-Wheeler transform to create a permanent, reusable index of the genome. Backtracking is used to conduct a quality-aware, greedy, randomized, depth-first search of all possible alignments based on the specified alignment parameters. Does not handle gapped alignments. Fast, memory efficient, and accurate for short reads of high quality (<50bp). Popular for short DNA-Seq reads and small RNA-Seq reads. (http://bowtie-bio.sourceforge.net/index.shtml)
Bowtie 22 (Version 2.2.5) - Uses a Burrows-Wheeler transform to create a permanent, reusable index of the genome. Alignment involves mapping seed sequences in an ungapped fashion and then performing a gapped extension. Supports a local alignment mode that "soft clips" alignments which do not align end-to-end. Unlike Bowtie, handles gapped alignments, ambiguous bases (N’s), and paired reads that do not align in a paired fashion. Fast, memory efficient, and accurate for longer reads (>50bp) with no upper limit on read length. Popular for DNA-seq reads and small RNA-Seq reads. (http://bowtie-bio.sourceforge.net/bowtie2/index.shtml)
BWA3,4 (Version 0.7.15) - Uses a Burrows-Wheeler transform to create an index of the genome. Handles gapped alignments and ambiguous bases (N’s). BWA-backtrack uses a backward search may be optimal for short reads (>70bp). BWA-MEM typically fastest and most accurate for longer reads, although BWA-SW may have better sensitivity when gapped alignments. Popular for DNA-seq variant calling pipelines, but not for RNA-seq as splicing is not taken into account. (http://bio-bwa.sourceforge.net/)
GSNAP5 (Version 2015-12-31(v8)) - A short read aligner (>14bp) using a successive constrained search, capable of handling splicing using either a probabilistic model or database. Built to handle SNPs in alignment. Good sensitivity but slower speed and higher memory usage. Popular for RNA-seq analysis. (http://research-pub.gene.com/gmap/)
HISAT26 (Version 2.1.0) - A fast and sensitive alignment program for mapping next-generation sequencing reads (both DNA and RNA) to a population of genomes. HISAT2 is a successor to TopHat2. (https://github.com/DaehwanKimLab/hisat2)
Isaac 27 (Version 15.07.16) - Gapped aligner that finds candidate mapping positions by matching 32-mers from the data to 32-mers from the reference, extending the candidate mappings to the whole read, and selecting the best mapping. Has utility for mappying DNA-Seq with good speed and accuracy but high memory usage. (https://github.com/Illumina/isaac2)
STAR8 (Version 2.6.1d) - Splice-aware aligner that utilizes novel sequential maximal mappable seed search capable of handling splice junctions. Seeds are subsequently stitched together by local alignment. Capable of handling long reads. Good speed and sensitivity for RNA-seq analysis but with high memory usage. (https://github.com/alexdobin/STAR)
TMAP9 (Version 5.0.0) - Integrates a set of aligners to (including modified BWA) to identify candidate mapping locations and performs alignment using Smith-Waterman algorithm. TMAP is optimized to handle variable length reads and error profiles generated by Ion Torrent data. (https://github.com/iontorrent/TMAP)
TopHat10 (Version 1.4.1 with Bowtie 1.0.0) - Two stage aligner that first utilizes Bowtie to map to a reference and subsequently unaligned reads are are mapped to a database of possible splice junctions. Popular for RNAseq analysis with solid performance, speed, and memory usage. (https://ccb.jhu.edu/software/tophat/index.shtml)
TopHat 211 (Version 2.1.0) - A newer version of TopHat that utlizes Bowtie2 and refined algorithms from Tophat to improve both speed and accuracy. Popular for RNAseq analysis with solid performance, speed, and memory usage. (https://ccb.jhu.edu/software/tophat/index.shtml)
Task Dialog
Selecting an aligner will open the task dialog (Figure 2). All aligners will have an index selection section where the genome build for the species of interest must be entered for Assembly and the Aligner Index must be specified. Aligner indexes provide a means to break apart the reference sequence for fast sequence matching, and can be created for the whole genome or for regions of interest in a Gene/Feature annotation file. Adding Reference Aligner Indexes or Adding Aligner Indexes based on an Annotation Model can be performed via Library File Management or built on the fly.
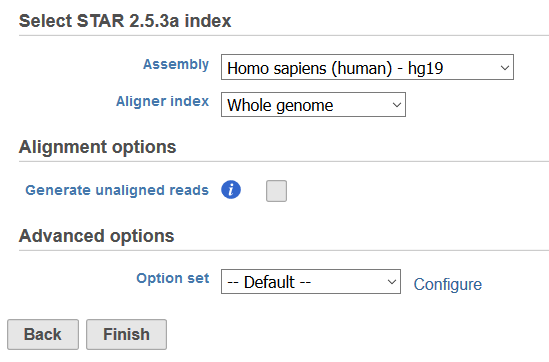
Figure 2. Example of an aligner task dialog for STAR
The Alignment options section is available for all aligners and includes the option to Generate unaligned reads. Selecting this option will create a new fastq file for each sample in the project that contains the reads that do not map during the alignment process.
In addition, some aligners have additional options specific to that tool. BWA allows for selection of the Alignment algorithm, including backtrack, MEM and SW (see BWA documentation). GSNAP has multiple options for Alignment mode (see GSNAP documentation). Both TopHat and TopHat2 have the option to select Fusion search (see Fusion Gene Detection).
The Advanced options section allows for the customization of option sets (see Option Set Management), which allows for the ability to specify parameters specific to each aligner. Default parameters are those specified by the developer of each aligner and parameter details found in the documentation for each aligner.
References
1. Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25.
2. Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357-359.
3. Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25(14):1754-1760.
4. Li H, Durbin R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics. 2010;26(5):589-595.
5. Wu TD, Nacu S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinforma Oxf Engl. 2010;26(7):873-881.
6. Kim D, Langmead B and Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nature Methods 2015
7. Raczy C, Petrovski R, Saunders CT, et al. Isaac: Ultra-fast whole genome secondary analysis on Illumina sequencing platforms. Bioinformatics. June 2013:btt314.
8. Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinforma Oxf Engl. 2013;29(1):15-21.
9. Torrent Suite User Documentation : Technical Note - TMAP Alignment (https://ts-pgm.epigenetic.ru/ion-docs/Technical-Note---TMAP-Alignment_9012907.html).
10. Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinforma Oxf Engl. 2009;25(9):1105-1111.
11. Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36.
Additional Assistance
If you need additional assistance, please visit our support page to submit a help ticket or find phone numbers for regional support.


| Your Rating: |
|
Results: |

|
25 | rates |
Overview
Content Tools
1 Comment
Melissa del Rosario
author: eseiser