What is Count feature barcodes?
Count feature barcodes is a tool for quantifying the number of feature barcodes per cell from CITE-Seq, cell hashing, or other feature barcoding assays to measure protein expression. The input for Count feature barcodes is FASTQ files.
Running Count feature barcodes
Count feature barcodes will run on any unaligned reads data node.
- Click the data node containing your unaligned reads containing feature barcodes
- Click the Quantification section in the toolbox
- Click Count feature barcodes
The task set up page allows you to configure the settings for your assay (Figure 1).
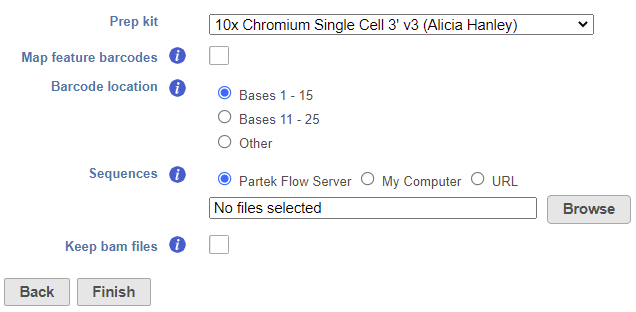 Figure 1. Count feature barcodes task set up page
Figure 1. Count feature barcodes task set up page
- Choose the Prep kit from the drop-down menu
For more details on adding Prep kits, please see our documentation on Trim tags. The prep kit should include cell barcode and unique molecular identifiers (UMIs) locations.
- Check Map feature barcodes box (optional)
This is only necessary for processing data from 10X Genomics' Feature Barcoding assay (v3+ chemistry), which utilizes BioLegend TotalSeq-B. For all other assays, leave this box unchecked.
- Choose the Barcode location
For BioLegend TotalSeq-A, choose bases 1-15. For BioLegend TotalSeq-B/C, choose bases 11-25. For other locations, select Custom and specify the start and stop positions.
- Choose a Sequences text file
This tab-delimited text file should have the feature ID in the first column and the nucleotide sequence in the second column. Do not include column headers. See Figure 2 for an example.
 Figure 2. Example of how the sequences tab-delimited text (.txt) file should be formatted
Figure 2. Example of how the sequences tab-delimited text (.txt) file should be formatted
- Check Keep bam files box (optional)
This option will retain the alignment BAM files instead of automatically deleting them when the task is complete. An extra Aligned reads output data node will be produced on the task graph. This option is unchecked by default to save on disk space.
- Click Finish to run
The output of Count feature barcodes is a Single cell counts data node.
How does Count feature barcodes work?
Count feature barcodes uses a series of tasks available independently in Partek Flow to process the input FASTQ files. The output files generated by these tasks are not retained in the Count feature barcodes output, with the exception of BAM files if Keep bam files is checked.
Trim tags identifies the UMI and cell barcode sequences. The Prep kit is specified using the Prep kit setting.
Trim bases trims the insert read to include only the feature barcode sequence. Trim bases is set to Both ends for Trim based on with the start and stop set by the Barcode location preference and the Min read length set to 1.
Bowtie is used to align the reads to the sequences specified in the Sequences text file. Bowtie is set to Ignore quality limit for the Alignment mode. Other settings are default.
Deduplicate UMIs consolidates duplicate reads based on UMIs. Deduplicate UMIs is set to Retain only one alignment per UMI.
Filter barcodes filters the cell barcodes to include cells and not empty droplets. Filter barcodes is set to Automatic.
Quantify barcodes counts the number of UMIs per cell for each feature in the Sequences file. Quantify barcodes uses default settings.
To perform these steps individually instead of using the Count feature barcodes task, you will need to generate a FASTA and GTF file containing the feature barcode IDs and sequences instead of a text file and build an index file for the Bowtie aligner.
For more information about library file management, please see Library File Management.
Additional Assistance
If you need additional assistance, please visit our support page to submit a help ticket or find phone numbers for regional support.


| Your Rating: |
|
Results: |

|
12 | rates |
Overview
Content Tools