The Deduplicate UMIs task identifies and removes reads mapped to the same chromosomal location with duplicate unique molecular identifiers (UMIs). The details of our UMI deduplication methods are outlined in the UMI Deduplication in Partek Flow white paper.
To invoke Deduplicate UMIs:
- Click an Aligned reads data node
- Click Post-alignment tools in the toolbox
- Click Deduplicate UMIs
The task configuration dialog content depends on whether you imported FASTQ files or BAM files into Partek Flow.
Configuring Deduplicate UMIs
Imported FASTQ
UMIs and barcodes are detected and recorded by the Trim tags task in Partek Flow. You can choose whether to retain only one alignment per UMI or not (Figure 1). The default will depend on which prep kit was used in the Trim tags task.
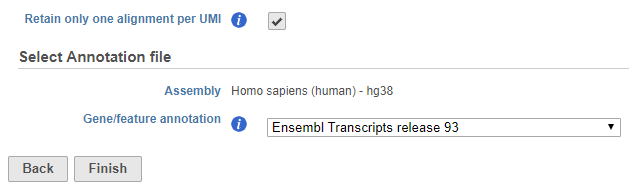
Figure 1. UMI deduplication dialog if UMIs and barcodes were processed by Trim tags
If you select Retain only one alignment per UMI, you will be asked to choose an assembly and gene/feature annotation file. The annotation file is used to check whether a read overlaps an exonic region. Only reads that have 50% overlap with an exon will be retained.
If you do not select Retain only one alignment per UMI, UMI deduplication will proceed without filtering to exonic reads. Other differences between the two options are outlined in the UMI Deduplication in Partek Flow white paper.
Imported BAM
Imported BAMs generated by other tools can be imported into Partek Flow and deduplicated by the software. Additional options are available in the task configuration dialog to allow you to specify the location of the UMI and cell barcode information typically stored in the BAM header. Specify the BAM header tags in the text fields. For example, when processing a BAM file produced by CellRanger 3.0.1, the BAM identifier tag for the UMI sequence is UR and the BAM identifier for the barcode sequence is CR (Figure 2).
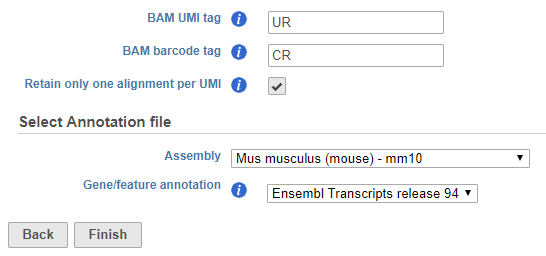
Figure 2. Specify the location of the BAM UMI and barcode tags
The option to Retain only one alignment per UMI is also available when starting from a BAM file.
Deduplicate UMIs task report
The Deduplicate UMIs task report includes a knee plot showing the number of deduplicated reads per barcode. This plot is used to filter the barcodes to include only barcodes corresponding to cells. For more information about using the knee plot to filter barcodes, please see the Cell Barcode QA/QC page. One difference between the Deduplication report and the Cell Barcode QA/QC report is that the Deduplication report gives the number of initial alignments and the number of deduplicated alignments for each sample (Figure 3). This indicates how many of your aligned reads were PCR duplicates and how many were unique molecules.
The initial number of cells is set by our automatic filter. You can set the filter manually by clicking on the plot or by typing a cutoff number in the Cells or Reads in cells text boxes. If there are multiple samples, each sample receives a plot and filters are set per sample.
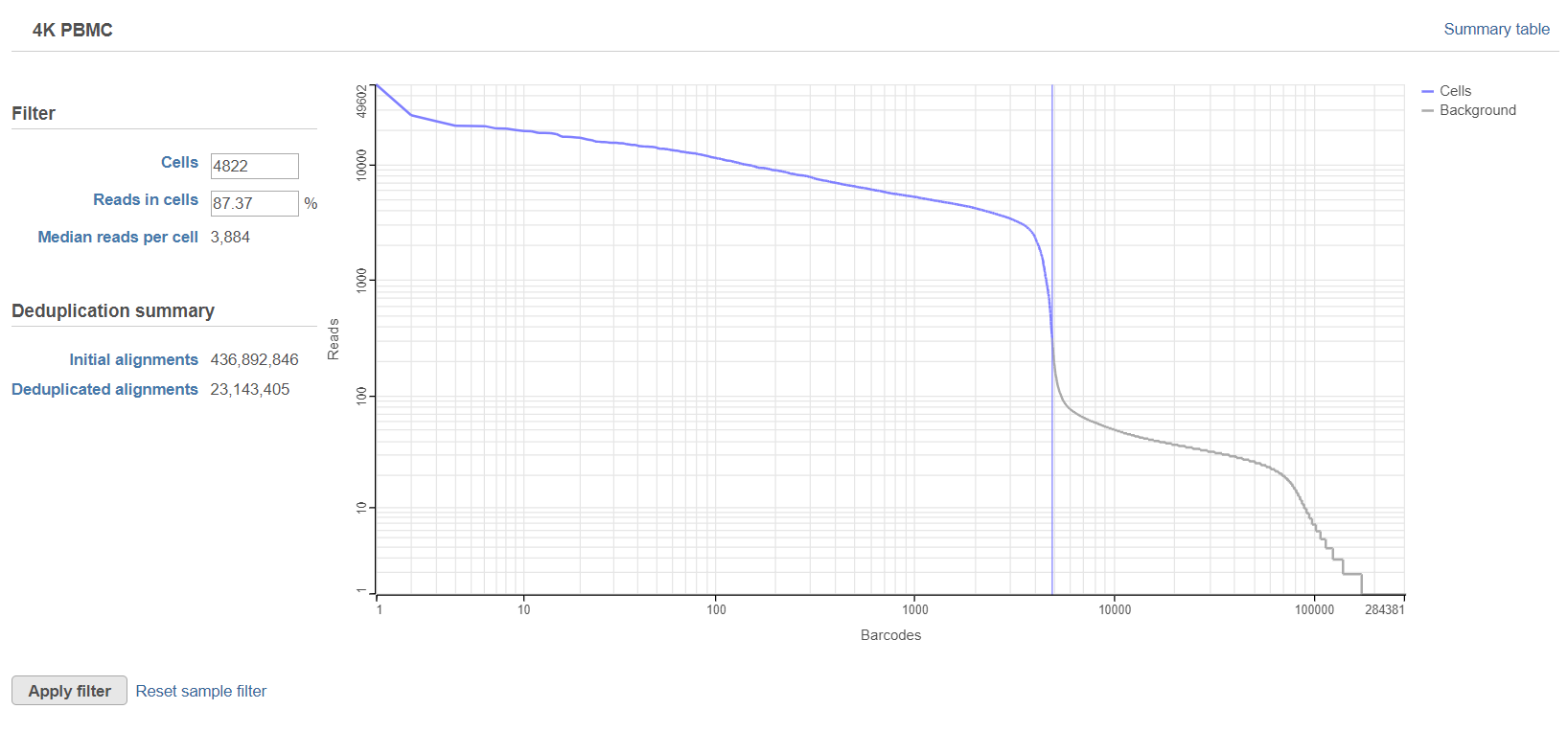
Figure 3. The deduplication report shows the number of UMIs per barcode
The number of cells, reads in cells, median reads per cell, number of initial alignments, and number of deduplicated alignments are listed for each sample in the summary table (Figure 4).
 Figure 4. Deduplication report summary table
Clicking Apply filter at either the knee plot or the summary table will run the Filter barcodes task and generate a Filtered reads data node.
Figure 4. Deduplication report summary table
Clicking Apply filter at either the knee plot or the summary table will run the Filter barcodes task and generate a Filtered reads data node.
To return to the knee plot, click Back to filter.
To reset the filters for all sample to the automatic cutoff, click Reset all filters.
Additional Assistance
If you need additional assistance, please visit our support page to submit a help ticket or find phone numbers for regional support.


| Your Rating: |
|
Results: |

|
29 | rates |
Overview
Content Tools