Partek Flow can import a wide variety of data types including raw counts, matrices, microarray, variant call files as well as unaligned and aligned NGS data.
- SFTP File Transfer Instructions
- Import single cell data
- Importing 10x Genomics Matrix Files
- Importing and Demultiplexing Illumina BCL Files
- Partek Flow Uploader for Ion Torrent
- Importing 10x Genomics .bcl Files
- Import a GEO / ENA project
The following file types are valid and will be recognized by the Partek Flow file browser.
|
|
|
In cases where paired end fastq data is present, files will also be automatically recognized and their paired relationship will be maintained throughout the analysis.
Matching on paired end files is based on file names: every character in both file names must match, except for the section that determines whether a file is the first or the second file. For instance, if the first file contains "_R1", "_1", "_F3", "_F5" in the file name, the second file must contain something in the lines with the following: "_R2", "_2", "_F5", "_F5-P2", "_F5-BC", "_R3", "_R5" etc. The identifying section must be separated from the rest of the filename with underscores or dots. If two conflicting identifiers are present then the file is treated as single end. For example, s_1_1 matches s_1_2, as described above. However, s_2_1 does not mate with s_1_2 and the files will be treated as two single-end files.
Apart from paired-end data, files with conventional filename suffixes that indicate that they belong to the same sample are consolidated. These suffixes include:
- Adapter sequences
- "_bbbbbb" followed by "_" or at the end of the file name, where each "b" is "A", "C", "G", or "T"
- Lane numbers
- "_L###" followed by "_" or at the end of the file name, where each "#" is a digit 0 to 9
Dates
in the form "####-##-##" preceded or followed by a period or underscore
Set number
of the form "_###" from the end
Navigating the file browser to transfer files to the server
The file browser is used to transfer files to the server so that these files can be added to a project for analysis. If you are importing a Bioproject from GEO/ENA or using URLs for data import, there is no need to transfer the files to the server.
To access the file browser and upload data to the server, use any of these options:
- access Transfer files
 on the Partek® Flow® homepage
on the Partek® Flow® homepage - within a project, after selecting the file type to transfer, using the transfer files link
 available within all file import options
available within all file import options - from the settings, go to Access management > Transfer files
Using the file browser to transfer files to the server:
- Click Transfer files
 to access the file browser
to access the file browser - Drag and drop or click My Device
 to add files from your machine
to add files from your machine - Click Browse
 to modify the Upload directory or create a new folder. The Upload directory
to modify the Upload directory or create a new folder. The Upload directory 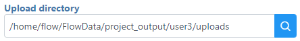 should be specified, known, and distinguishable for project file management. You will return to this directory and access the files to import them into a project
should be specified, known, and distinguishable for project file management. You will return to this directory and access the files to import them into a project - To continue to add more files use + Add more
 in the top right corner. To cancel the process select Cancel in the top left corner
in the top right corner. To cancel the process select Cancel in the top left corner - Click Upload
 to complete the file upload
to complete the file upload - Do not exit the browser tab or let the computer go to sleep or shut down until the transfer has completed
File size displayed in the table is binary format, not decimal format (e.g. GB displayed in the table is gigibyte not gigabyte. 1 gigibyte is 1,073,741,824 bytes. 1 gigabyte is 1,000,000,000 bytes. 1 gigibyte is 1.074 gigabytes).
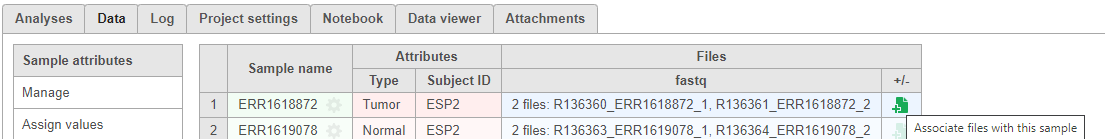
Associate fastq files for multi-omic data
There are projects with more than one file type, such as single cell multi-omic assays that generate protein and RNA together. In these cases, files need to be associated with each other if starting in fastq format. If we start with processed data, there is no need to associate these files.
- Define the type of data the file represents when importing files into the project.
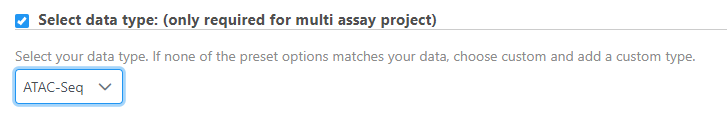
If this step is skipped, the data type can be changed after import by right clicking the data node.
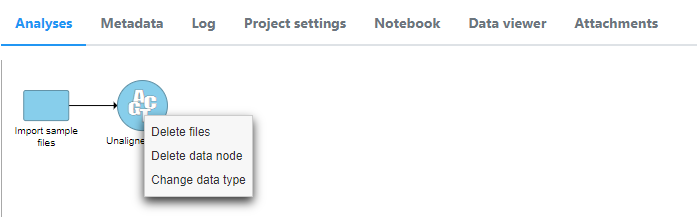
- After importing both types of data, associate fastqs with the already imported data. (e.g. associate RNA fastqs with ATAC fastqs already imported into Partek Flow.
Additional Assistance
If you need additional assistance, please visit our support page to submit a help ticket or find phone numbers for regional support.


| Your Rating: |
|
Results: |

|
35 | rates |
Partek
Overview
Content Tools