RNA-Seq uses the number of sequencing reads per gene or transcript to quantify gene expression. Once reads are aligned to a reference genome, we need to assign each read to a known transcript or gene to give a read-count per transcript or gene.
- Click the Aligned reads data node
- Click Quantification in the task menu
We will use Partek E/M to quantify reads to an annotation model in this tutorial. For more information about the other quantification options, please see the Quantification user guide.
- Click Quantify to an annotation model (Partek E/M) (Figure 1)
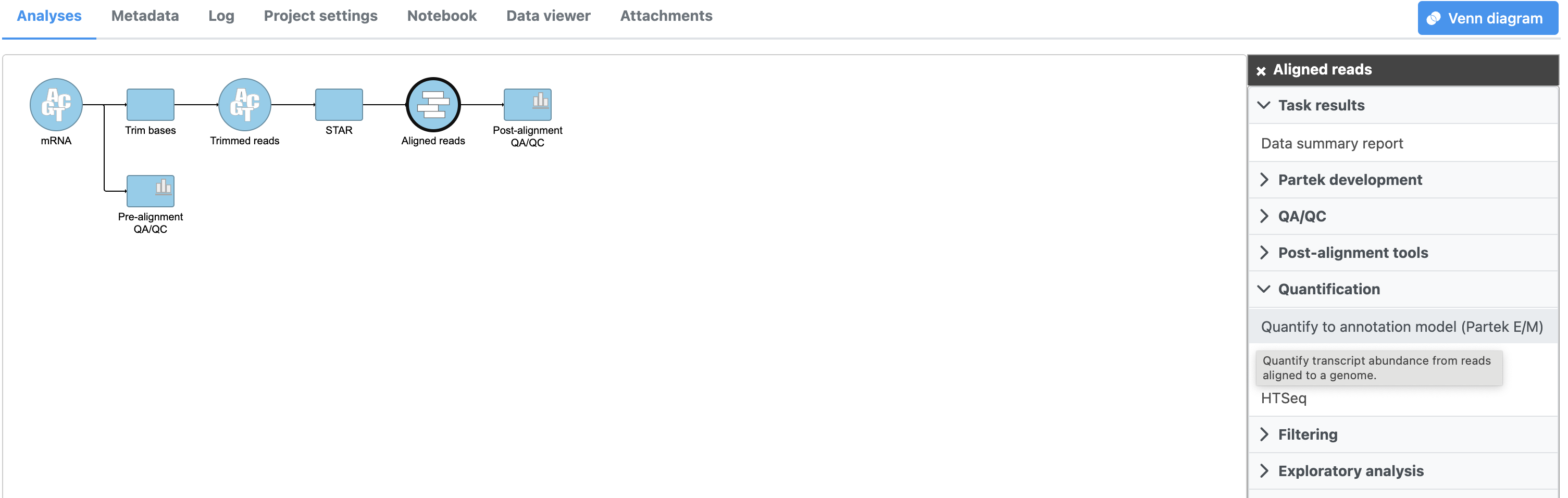 Figure 1. Invoking Quantify to an annotation model (Partek E/M)
We will use the default options for quantification. To learn more about the different options, please see the Quantify to annotation model (Partek E/M) user guide or mouse over the
Figure 1. Invoking Quantify to an annotation model (Partek E/M)
We will use the default options for quantification. To learn more about the different options, please see the Quantify to annotation model (Partek E/M) user guide or mouse over the  next to each option.
next to each option.
- Choose the latest RefSeq Transcripts 95 annotation from the Gene/feature annotation drop-down menu (you may need to download it first, via Library File Management)
- Click Finish (Figure 2)
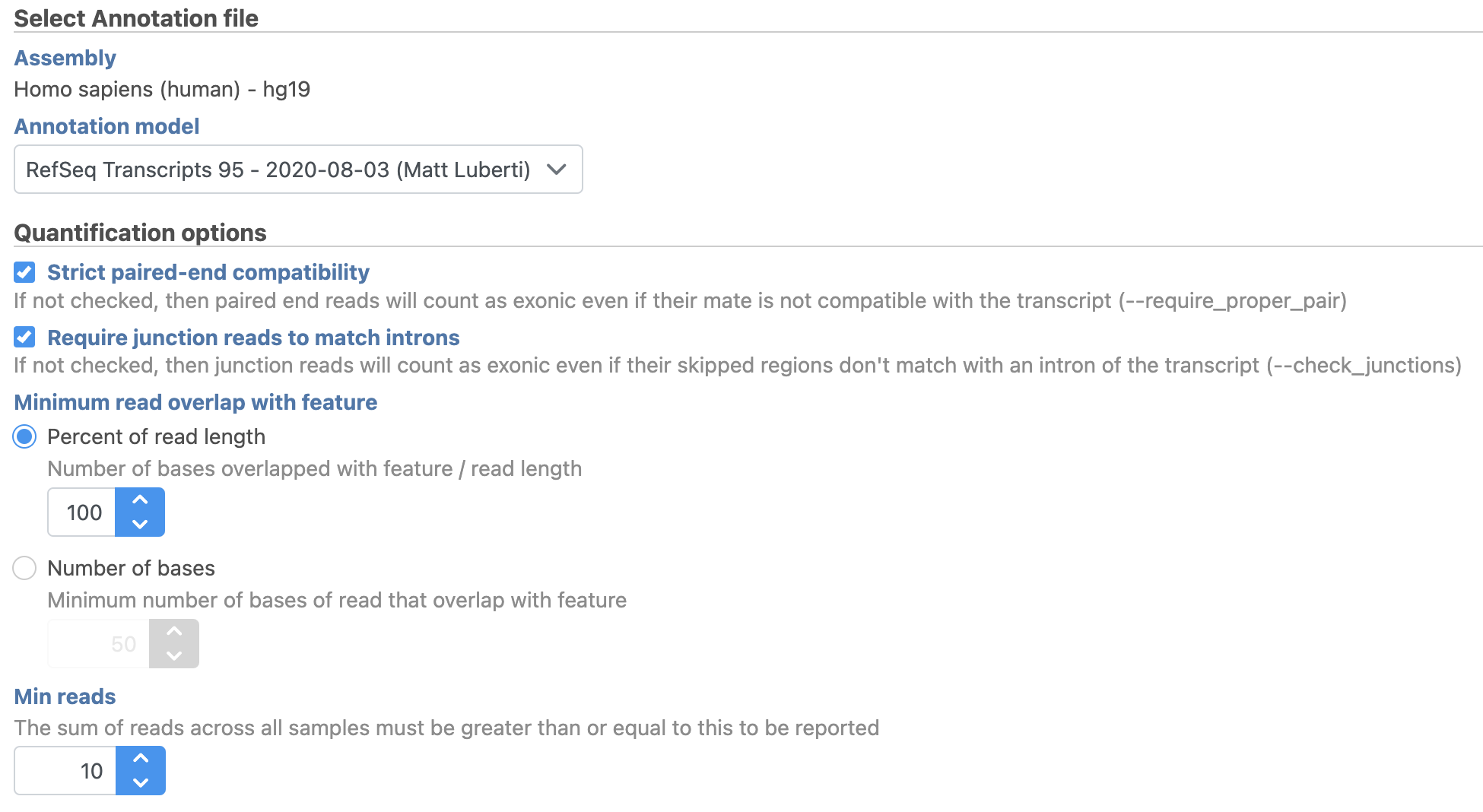
Figure 2. Configuring Quantify to annotation model (Partek E/M)
The Quantify to annotation model task node outputs two data nodes, Gene counts and Transcript counts (Figure 3).
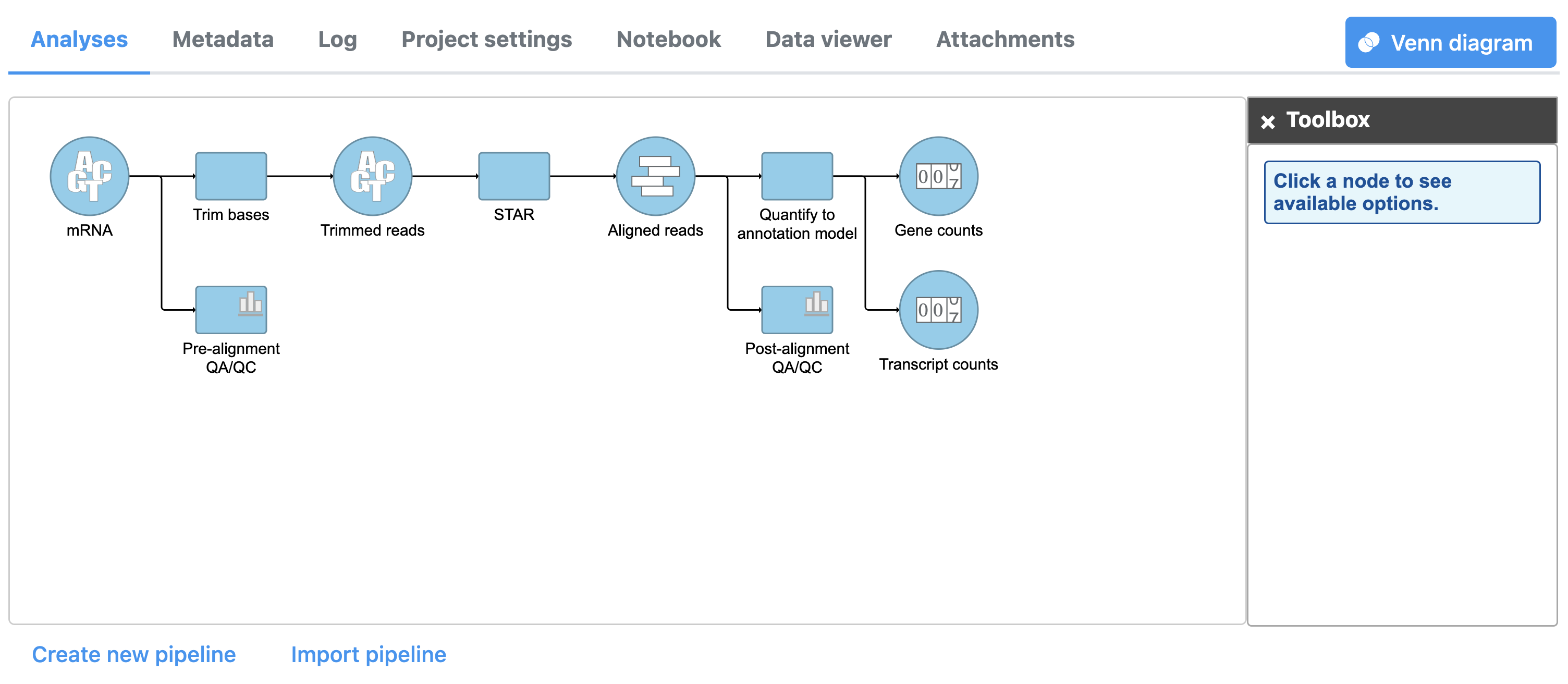
Figure 3. Gene counts and Transcript counts data nodes
To view the results of quantification, we can select either data node output.
- Double-click the Gene counts data node to view the task report
The task report details the number of reads within exons, introns, and intergenic regions. For detailed information about the quantification results, see the Quantify to annotation model (Partek E/M) user guide.
Additional Assistance
If you need additional assistance, please visit our support page to submit a help ticket or find phone numbers for regional support.


| Your Rating: |
|
Results: |

|
30 | rates |
Overview
Content Tools