When the reads are aligned to a genome reference, e.g. hg38, the quantification is performed on transcriptome, you need to provide the annotation model file of the transcriptome.
Quantification dialog
If the alignment was generated in Partek Flow, the genome assembly will be displayed as text on the top of the page (Figure 1), you do not have the option to change the reference.
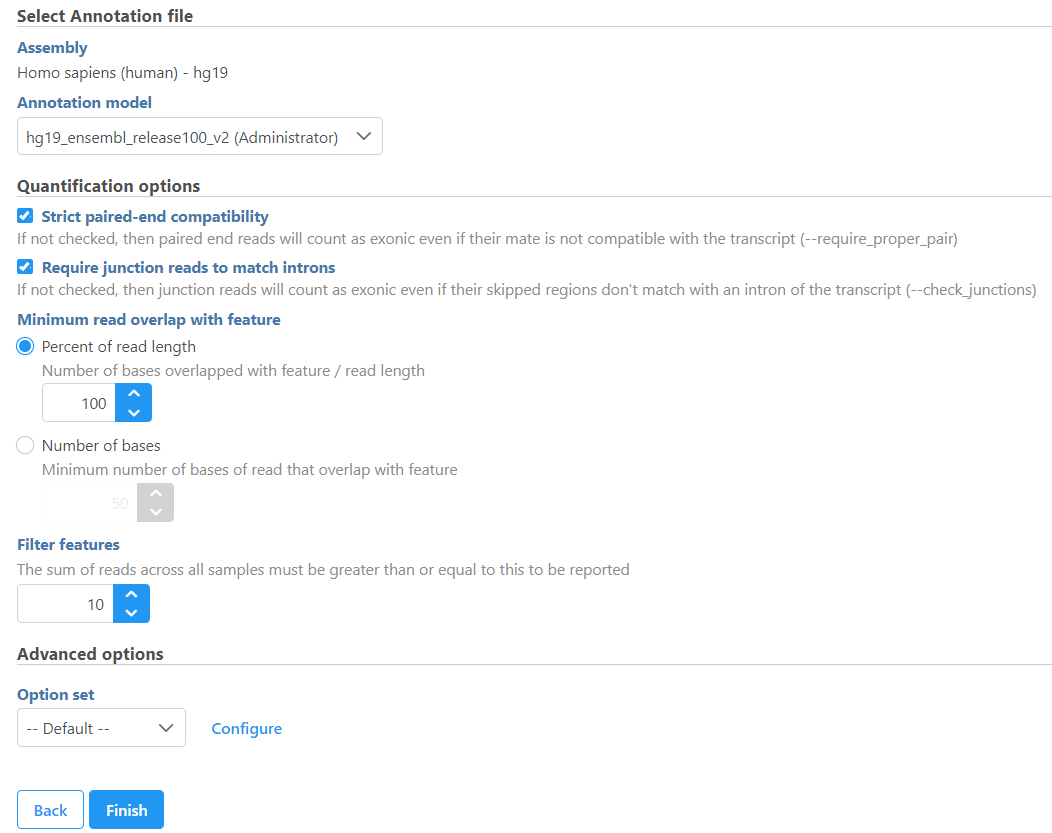
Figure 1. Quantify to annotation model(Partek E/M) dialog
If the bam file is imported, you need to select the assembly with which the reads were aligned to, and which annotation model file you will use to quantify from the drop-down menus (Figure 2).
Figure 2. Specify the genome assembly with which the bam files are generated from and transcriptome annotation from the drop-down menu
In the Quantification options section, when the Strict paired-end compatibility check button is selected, paired end reads will be considered compatible with a transcript only if both ends are compatible with the transcript. If it is not selected, reads with only one end have alignment that is compatible with the transcript will also be counted for the transcript .
If the Require junction reads to match introns check button is selected, only junction reads that overlap with exonic regions and match the skipped bases of an intron in the transcript will be included in the calculation. Otherwise, as long as the reads overlap within the exonic region, they will be counted. Detailed information about read compatibility can be found in the Understanding Reads white paper.
Minimum read overlap with feature can be specified in percentage of read length or number of bases. By default, a read has to be 100% within a feature. You can allow some overhanging bases outside the exonic region by modifying these parameters.
Filter features option is a filter for minimum reads, by default only the features whose sum of the reads across all samples that are greater than or equal to 10 will be reported. To report all the features in the annotation file, set the value to 0.
Some library preparations reverse transcribe the mRNA into double stranded cDNA, thus losing strand information. In this case, the total transcript count will include all the reads that map to a transcript location. Others will preserve the strand information of the original transcript by only synthesizing the first strand cDNA. Thus, only the reads that have sense compatibility with the transcripts will be included in the calculation. We recommend verifying with the data source how the NGS library was prepared to ensure correct option selection.
In the Advanced options, in Configure dialog, at Strand specificity field, forward means the strand of the read must be the same as the strand of the transcript while reverse means the read must be the complementary strand to the transcript (Figure 3). The options in the drop-down list will be different for paired-end and single-end data. For paired-end reads, the dash separates first- and second-in-pair, determined by the flag information of the read in the BAM file. Briefly, the paired-end Strand specificity options are:
- No: Reads will be included in the calculation as long as they map to exonic regions, regardless of the direction
- Auto-detect: The first 200,000 reads will be used to examine the strand compatibility with the transcripts. The following percentages are calculated on paired-end reads:
- (1) If (first-in-pair same strand + second-in-pair same strand)/Alignments examined > 75%, Forward-Forward will be specified
- (2) If (first-in-pair same strand + second-in-pair opposite strand)/Alignments examined > 75%, Forward-Reverse will be specified
- (3) If (first-in-pair opposite strand + second-in-pair same strand)/Alignments examined > 75%, Reverse-Forward will be specified
- (4) If neither of the percentages exceed 75%, No option will be used
- Forward - Reverse: this option is equivalent to the --fr-secondstrand option in Cufflinks [1]. First-in-pair is the same strand as the transcript, second-in-pair is the opposite strand to the transcript
- Reverse - Forward: this option is equivalent to --fr-firststrand option in Cufflinks. First-in-pair is the opposite strand to the transcript, second-in-pair is the same strand as the transcript. The Illumina TruSeq Stranded library prep kit is an example of this configuration
- Forward - Forward: Both ends of the read are matching the strand of the transcript. Generally colorspace data generated from SOLiD technology would follow this format
The single-end Strand specificity options are:
- No: same as for paired-end reads
- Auto-detect: same as for paired-end reads. All single-end reads are treated as first-in-pair reads
- Forward: this option is equivalent to the --fr-secondstrand option in Cufflinks. The single-end reads are the same strand as the transcript
- Reverse: this option is equivalent to --fr-firststrand option in Cufflinks. The single-end reads are the opposite strand to the transcript. The Illumina TruSeq Stranded library prep kit is an example of this configuration
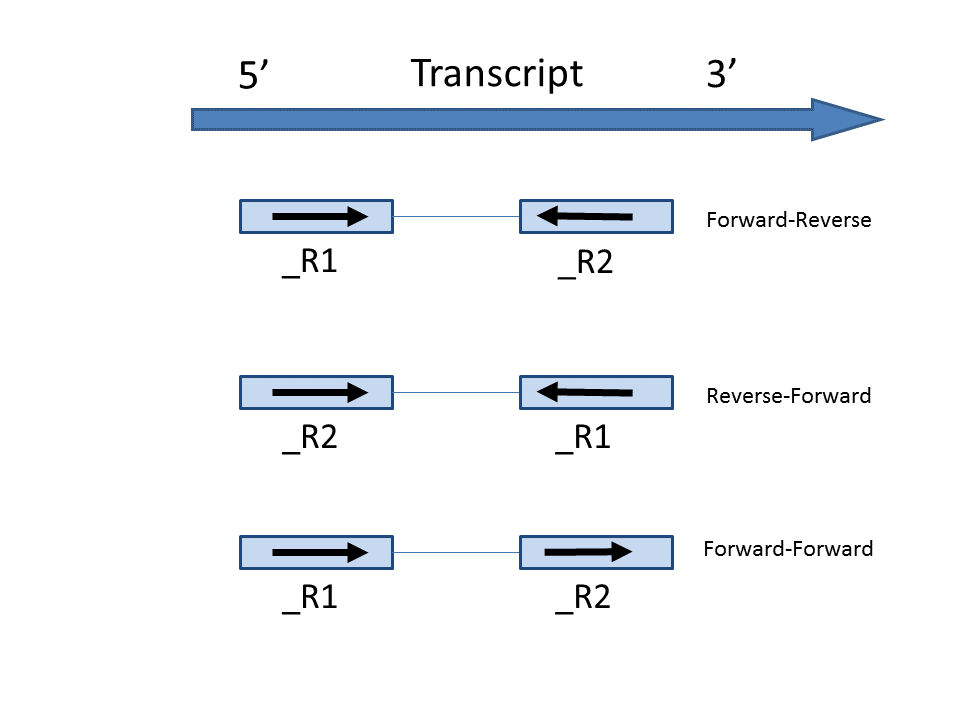
Figure 3. Illustration of the three types of strand specific assays on paired end reads. _R1 and _R2 means read first-in pair and second-in-pair respectively. Arrows indicate strand directions.
If the Report unexplained regions check button is selected, an additional report will be generated on the reads that are considered not compatible with any transcripts in the annotation provided. Based on the Min reads for unexplained region cutoff, the adjacent regions that meet the criteria are combined and region start and stop information will be reported.
In the annotation file, there might be multiple features in the same location, or one read might have multiple alignments, so the read count of a feature might not be an integer. Our white paper on the Partek E/M algorithm has more details on Partek’s implementation the E/M algorithm initially described by Xing et al. [1]
Quantify to annotation model (Partek E/M) output
Depending on the annotation file, the output could be one or two data nodes. If the annotation file only contains one level of information, e.g. miRNA annotation file, you will only get one output data node. On the other hand, if the annotation file contains gene level and transcript level information, such as those from the Ensembl database, both gene and transcript level data nodes will be generated. If two nodes are generated, the Task report will also contain two tabs, reporting quantification results from each node. Each report has two tables. The first one is a summary table displaying the coverage information for each sample quantified against the specified transcriptome annotation (Figure 4).
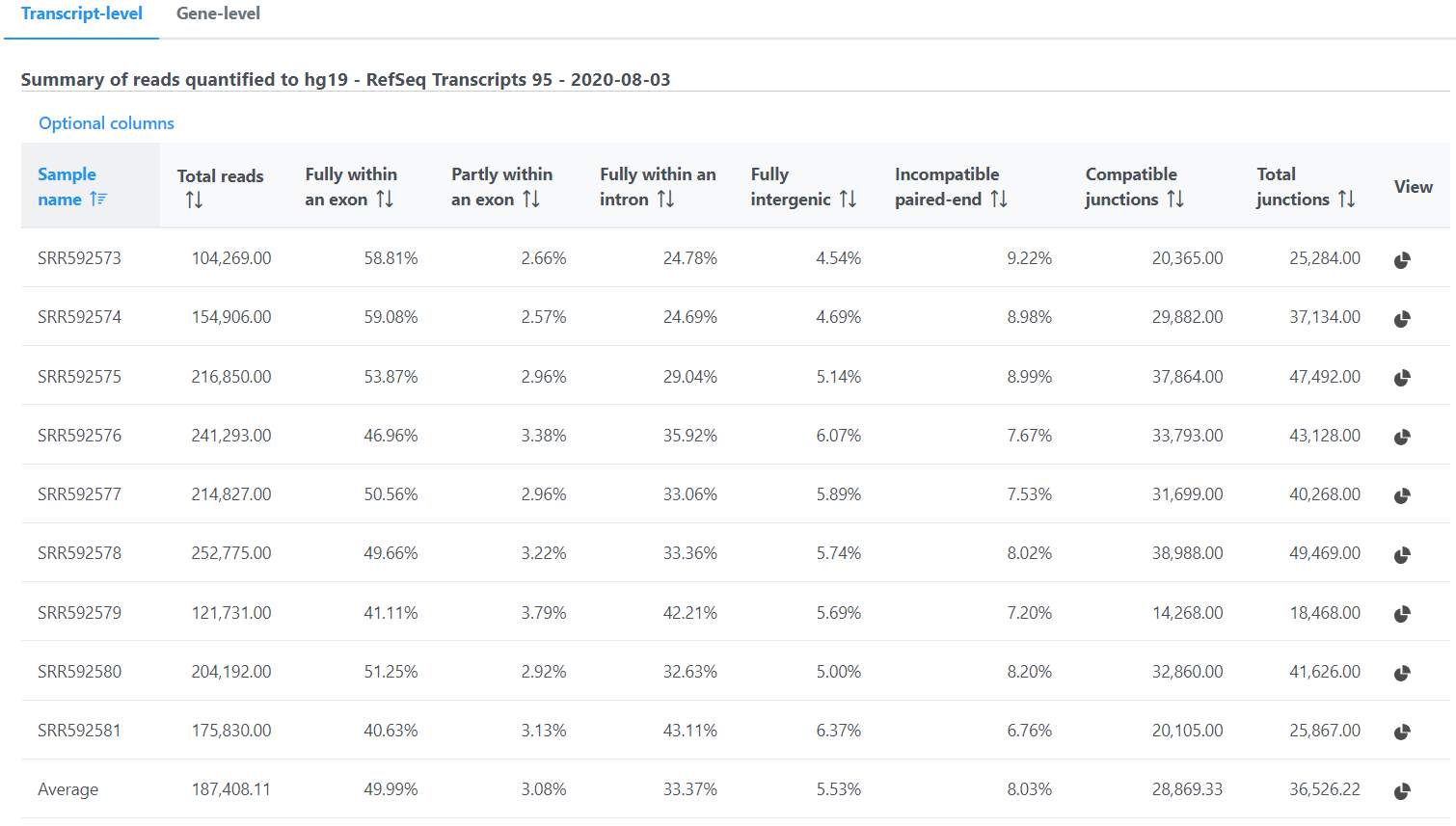
Figure 4. Summary of raw reads mapping to genes based on the RefSeq annotation file provided. Note that the Gene-level tab is selected.
The second table contains feature distribution information on each sample and across all the samples, number of features in the annotation model is displayed on the table title (Figure 5).
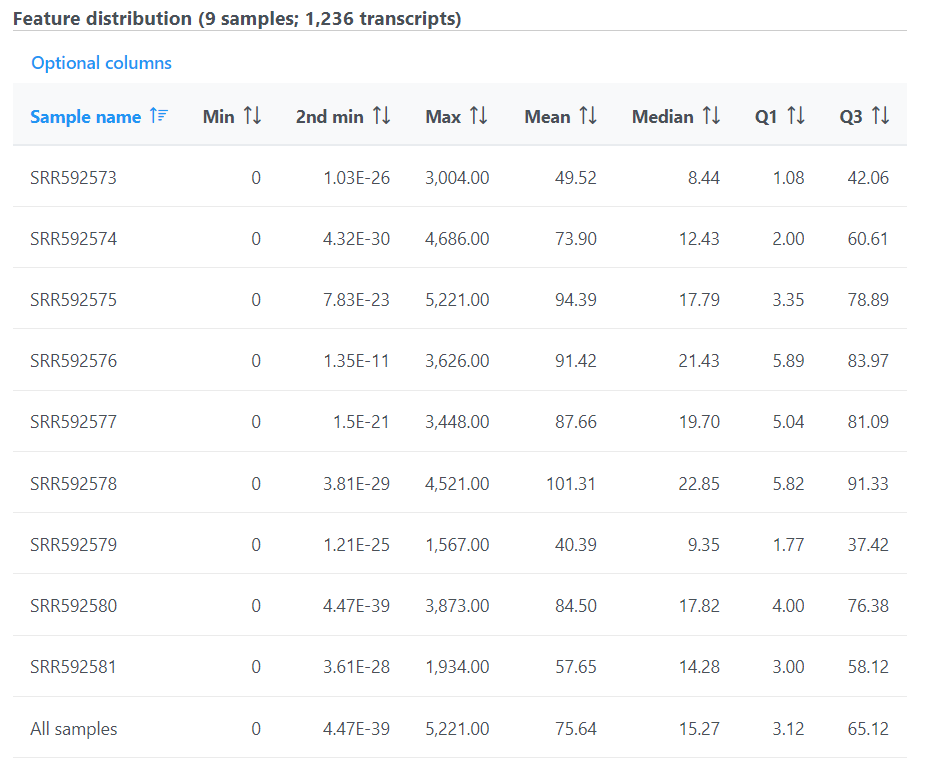
Figure 5. Summary of feature distribution statistics
The bar chart displaying the distribution of raw read counts is helpful in assessing the expression level distribution within each sample. The X-axis is the read count range, Y axis is the number of features within the range, each bar is a sample. Hovering your mouse over the bar displays the following information (Figure 6):
- Sample name
- Range of read counts, “[ “represent inclusive, “)” represent exclusive, e.g. [0,0] means 0 read counts; (0,10] means the range is greater than 0 count but less than and equal to 10 counts.
- Number of features within the read count range
- Percentage of the features within the read count range

Figure 6. Bar chart on distribution of raw read counts in each sample
The coverage breakdown bar chart is a graphical representation of the reads summary table for each sample (Figure 7)
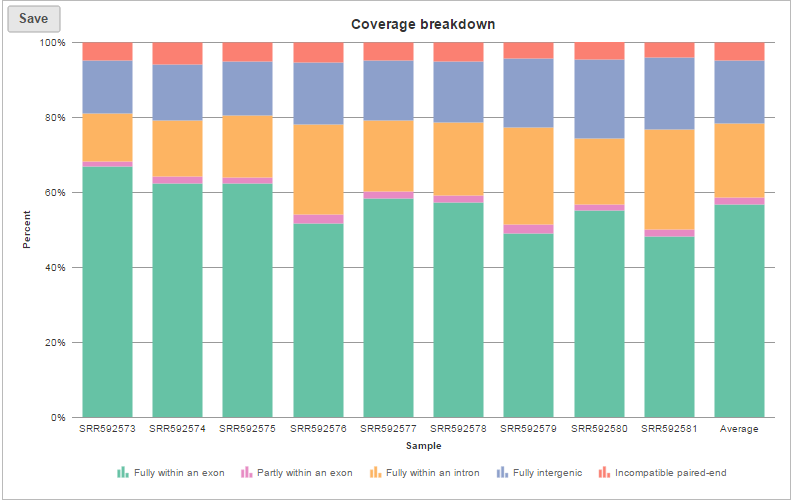
Figure 7. Coverage breakdown bar chart, it is a graphical presentation of summary table on raw reads mapping to transcription based on the annotation file provided
In the box-whisker plot, each box is a sample on X-axis, the box represents 25th and 75th percentile, the whiskers represent 10th and 90th percentile, Y-axis represents the feature counts, when you hover over each box, detailed sample information is displayed (Figure 8).
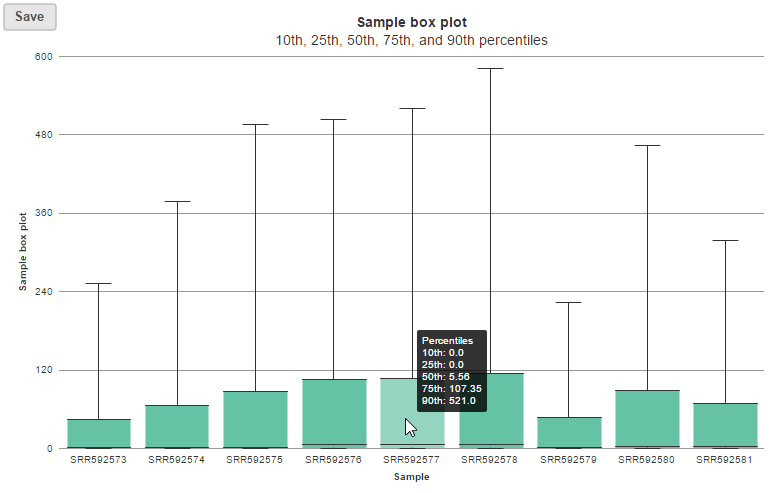
Figure 8. Box-whisker plot on read count distribution in each sample, when mouse over a box, detailed information on the box is displayed.
In sample histogram, each line represents a sample and the range of read counts are divided into 20 bins. Clicking on a sample in the legend will hide the line for that specific sample. Hovering over each circle displays detailed information about the sample and that specific bin (Figure 9). The information includes:
- Sample name
- Range of read counts, “[ “represent inclusive, “)” represent exclusive
- Number of features within the read count range in the sample
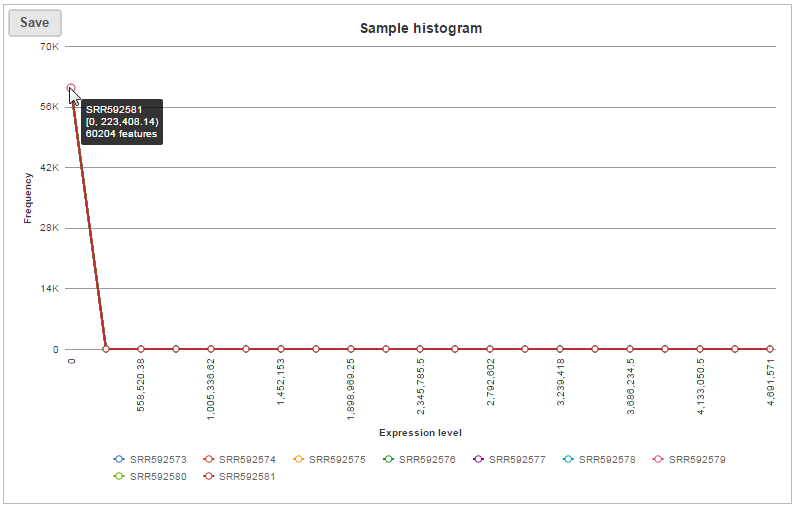
Figure 9. Sample histogram plot, when mouse over each circle, detailed information is displayed
The box whisker and sample histogram plots are helpful for understanding the expression level distribution across samples. This may indicate that normalization between samples might be needed prior to downstream analysis. Note that all four visualizations are disabled for results with more than 30 samples.
The output data node contains raw reads of each sample on each feature (gene or transcript or miRNA etc. depends on the annotation used). When click on a output data node, e.g. transcript counts data node, choose Download data on the context sensitive menu on the right, the raw reads of transcripts can be downloaded in three different format (Figure 10):
Partek Genomics Suite project format: it is a zip file, do not manually unzip it, you can choose File>Import>Zipped project in Partek Genomics Suite to import the zip file into PGS.
Features on columns and Features on rows format: it is a .txt file, you can open the text file in any text editor or Microsoft Excel. For Features on columns format, samples will be on rows. For Features on rows format, samples will be at columns.
 Figure 10. Download quantification output data dialog
Figure 10. Download quantification output data dialog
References
- Xing Y, Yu T, Wu YN, Roy M, Kim J, Lee C. An expectation-maximization algorithm for probabilistic reconstructions of full-length isoforms from splice graphs. Nucleic Acids Res. 2006; 34(10):3150-60.
Additional Assistance
If you need additional assistance, please visit our support page to submit a help ticket or find phone numbers for regional support.


| Your Rating: |
|
Results: |

|
43 | rates |
Overview
Content Tools
1 Comment
Melissa del Rosario
author: wxw