This white paper explains some basic concepts related to alignment and quantification tasks in Partek Flow. The term “alignment” describes the process of finding the position of a sequencing read on the reference genome, while quantification refers to assigning already aligned reads to transcripts based on specified transcripts/gene annotation.
Number of Reads and Alignments
Aligned data node contains reads that have been aligned, counting each read once even if it has multiple alignments. Processing of paired-end reads needs further elaboration: a paired-end read will be counted as one read only if both ends align to the genome, no matter how many alignments each end has (Figure 1). If one end of a paired-end read is not aligned, the read will be considered unaligned and will be discarded for the downstream analysis.
As reads can be aligned to more than one location, the number of alignments may be greater than the number of reads; since some reads may be unaligned, the number of alignments may be less than the number of reads in the bam/sam file (Figure 1).
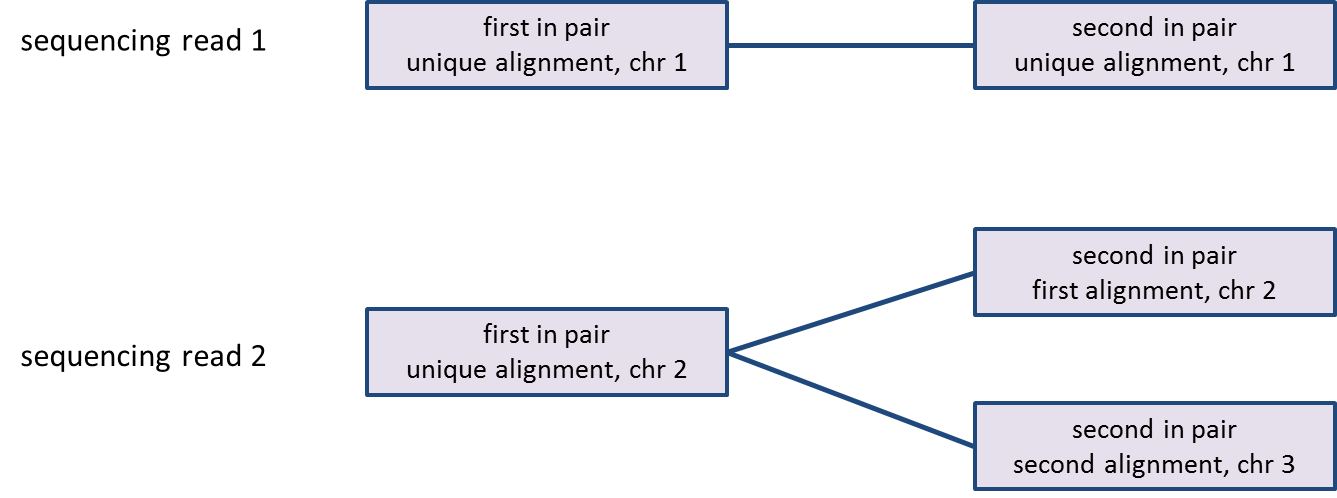
Figure 1. Counting reads and alignments for paired-end reads. Sequencing read 1 is imported as one paired-end read, with two alignments. Sequencing read 2 is imported as one paired-end read, with three alignments
Partek Flow shows the number of alignments as well as the aligned reads percentage per sample in the post alignment QC report.
Quantification: Assigning Reads to Transcripts
This step maps the aligned reads to transcripts using a modified E/M algorithm; detailed information about this algorithm can be found in the white paper RNA-Seq quantification.
Given a list of transcripts, each read can be mapped to one of four classes: exonic, partially overlaps exon, intronic, or between genes (Figure 2). Generally, “exonic” means the read came from a sequence present in the mature mRNA, while “intronic” means the read overlaps a gene, but aligns to the portion of the sequence not present in the mature mRNA. Finally, “between genes” reads came from a region outside of any gene. Detailed definitions of all four types can be found below.
The read assignments are labeled within the context of the chosen transcriptome database. If a read maps to multiple locations, then if any one of the alignments are exonic, the read is labeled “exonic”, and the same goes for partially overlaps exon, intronic and intergenic. In the other words, exonic takes precedence over partially overlaps exon, partially overlaps exon takes precedence over intronic, and intronic takes precedence over intergenic.
- Exonic: A read is labeled exonic if any one of its alignments is completely contained within the respective exon as defined by the database (read 1) ,i.e., even if there is a single base shift of the read relative to the exon, the read will not be called exonic but will fall in the category ‘partially overlaps exon’. If the alignments are strand specific, then the strand of the alignment must also agree with the strand of the transcript.
- Partially overlaps exon: A read is assigned to partially overlap an exon if any of its alignments overlaps an exon, but at least partly (one base-pair or more) maps out of the exon (read 3, read 4).
- Intronic: A read is labeled intronic if any one of its alignments maps completely within an intron (read 2), but none of the alignments are exonic (either fully or in part). If the alignments are strand-specific, then the strand of the alignment must also agree with the strand of the transcript.
- Reads between genes: A read is labeled ‘between genes’ if none of its alignments overlap a gene (read 5).
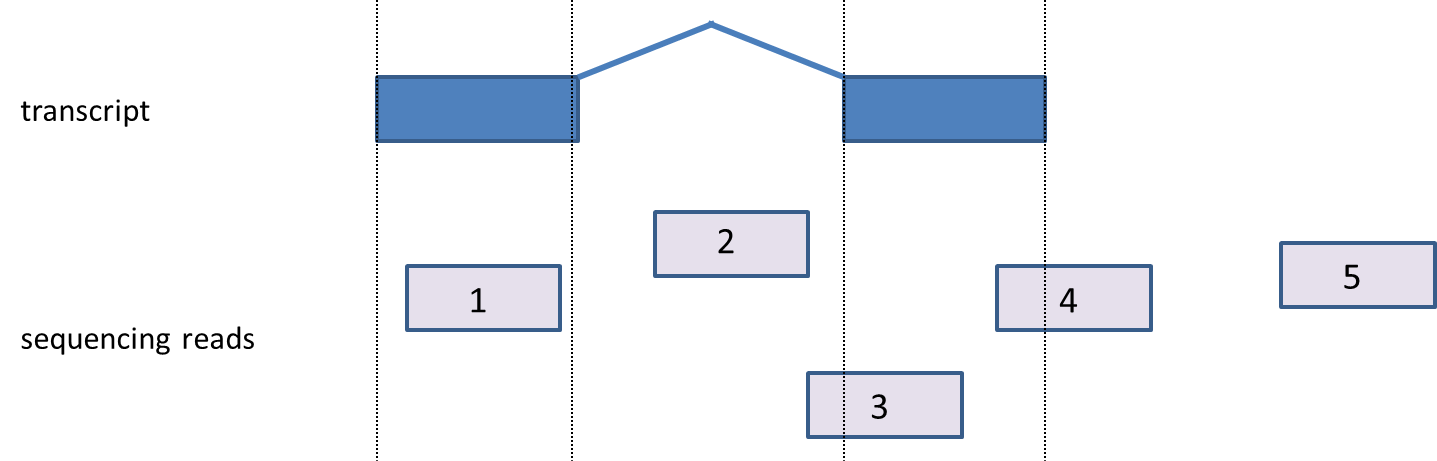
Figure 2. Mapping reads to transcripts. A transcript (blue) contains exonic (boxes) and intronic regions (the line joining the boxes). Sequencing reads (light blue) are assigned according to the positions they map to. 1: exonic (fully overlaps an exon), 2: intronic (fully contained within an intron), 3 & 4: partially overlap exon, 5: between genes
Read Compatibility
A read will be assigned to a transcript only when it is compatible. A compatible read is a read that fits the transcript model from the chosen annotation database. Compatible reads must be an exonic read (fully mapped to exon); however not all the exonic reads are compatible with a transcript, e.g., in paired end reads, both end reads have to be exonic as well as they both have to map to the same transcript; if they mapped to two different transcript or different chromosome, they are not compatible with a transcript.
Reads that are partially mapped exon reads, intronic reads, intergenic reads are incompatible with any transcript. Incompatible reads do not contribute to gene or transcript level read counts. Several single-end and paired-end scenarios will be discussed in the following sections.
Single-End Scenarios
A single-end read that is considered “compatible” would be any read that overlaps the exon 100% as described above in the paragraph on “exonic” reads.
If a single-end read includes an exon to exon junction, the region skipped in the read must be consistent with an intron in that transcript. If it is not, the read will be incompatible.
For example, with an alignment that begins at base 100 and has a cigar score of 5M 10N; 5M, the M means alignment Match and the N means skipped bases (junction). To be compatible, bases 100-104 must be exonic, bases 105-115 must be intronic and bases 116-120 must be exonic.
Paired-End Data Scenarios
The handling of paired-end data is a bit more complex. There can be zero or more alignments associated with the first-in-pair read as well as zero or more alignments associated with the second-in-pair read. Each of the alignments associated with a paired-end read is considered compatible/incompatible with overlapping transcripts using the same rules which apply to alignments associated with single-end reads. A paired-end read will be considered compatible if it contains any pair of alignments where an alignment from the first-in-pair read is compatible with a transcript and an alignment from the second-in-pair read is compatible with the same transcript. Consequently, if only one of the ends is compatible with the transcript, the read is counted as incompatible. Similar to that, if a paired-end read has a junction that is not consistent with the intron in the transcript, the read will be incompatible.
Considering genes with multiple transcripts, a read can be both counted as compatible for some transcripts, as well as counted incompatible for other transcripts of the same gene (Figure 3). Please note that this concept holds for single-end reads as well.
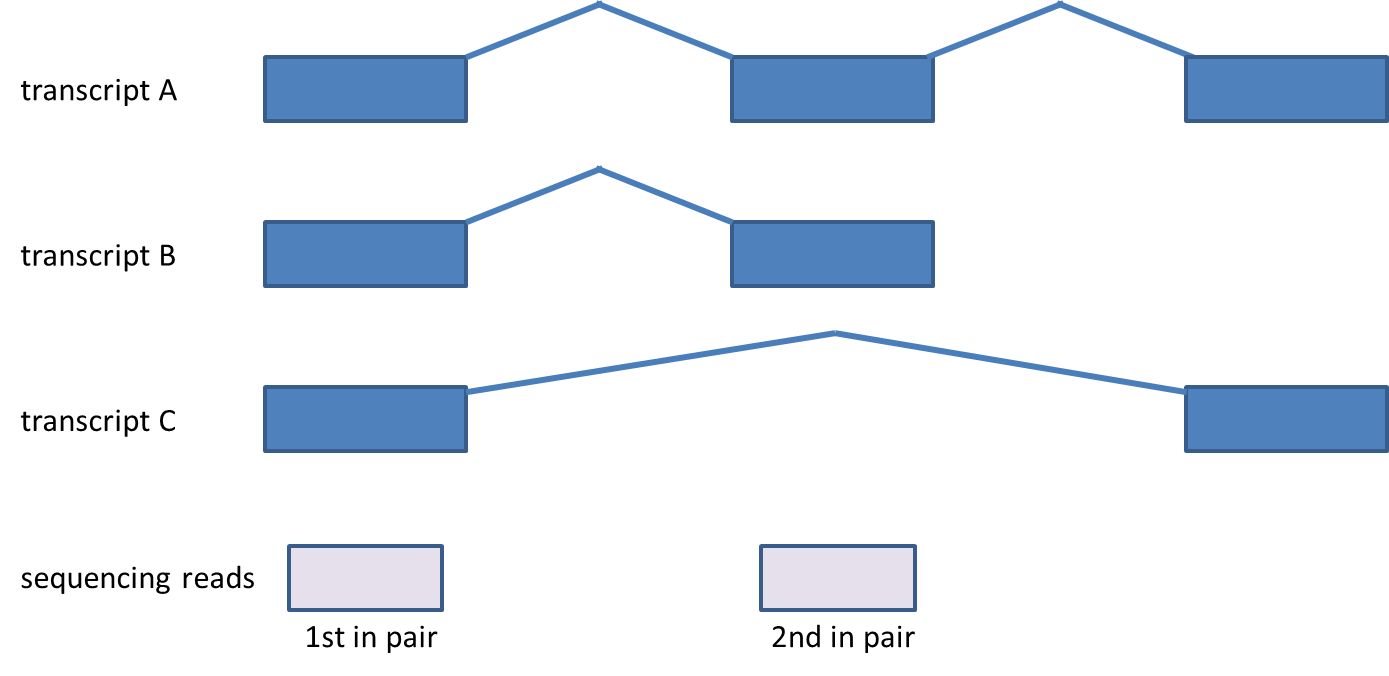
Figure 3. Compatibility of reads corresponding to genes with multiple transcripts. First in pair maps to all three transcripts, while second maps to transcripts A and B. The paired-end read is compatible with transcripts A and B, but is not compatible with the transcript C. Although the picture shows paired-end reads, the same rules apply for single-end reads
Furthermore, all the reads that have at least one alignment contribute to the total number of aligned reads.
Unexplained regions
The unexplained regions portion of quantification considers any read that is considered “not compatible” with all transcripts. It is basically a 3 step process:
- Filter down to reads that are not compatible with the transcript model.
- Call regions that have at least an average of 5 reads per position.
- Combine adjacent regions and report the result.
Since we are filtering down to reads that are incompatible with the transcript model, therefore during the combine adjacent regions step, only incompatible reads will be combined.
When interpreting the unexplained reads, you should have in mind that these are actually the reads not compatible with the applied transcript model. By changing the model, some reads will become compatible and thus will not be labeled “unexplained” any more. Figure 6 shows such an example. The depicted regions map just downstream of the human LONP2 gene as defined by RefSeq and are hence flagged as unexplained. However, by overlaying the AceView transcripts, it is apparent that mapping to AceView would yield a different result.

Figure 4. Regions track in the Partek Genome Viewer. Regions (colored boxes) detected in the four samples contain sequencing reads that are not compatible with the RefSeq database (upper transcripts track) but are compatible with at least one of the exons of the same gene defined by the AceView database (lower transcripts track)
Additional Assistance
If you need additional assistance, please visit our support page to submit a help ticket or find phone numbers for regional support.


| Your Rating: |
|
Results: |

|
38 | rates |
Overview
Content Tools
1 Comment
Melissa del Rosario
author: ilukic