With our reads trimmed, we now have high-quality reads for each sample. The next step is to align the reads to a reference genome. Alignment matches each of the short sequencing reads to a location in the reference genome.
- Click Trimmed reads
- Click Aligners in the task menu to display available aligners (Figure 1)
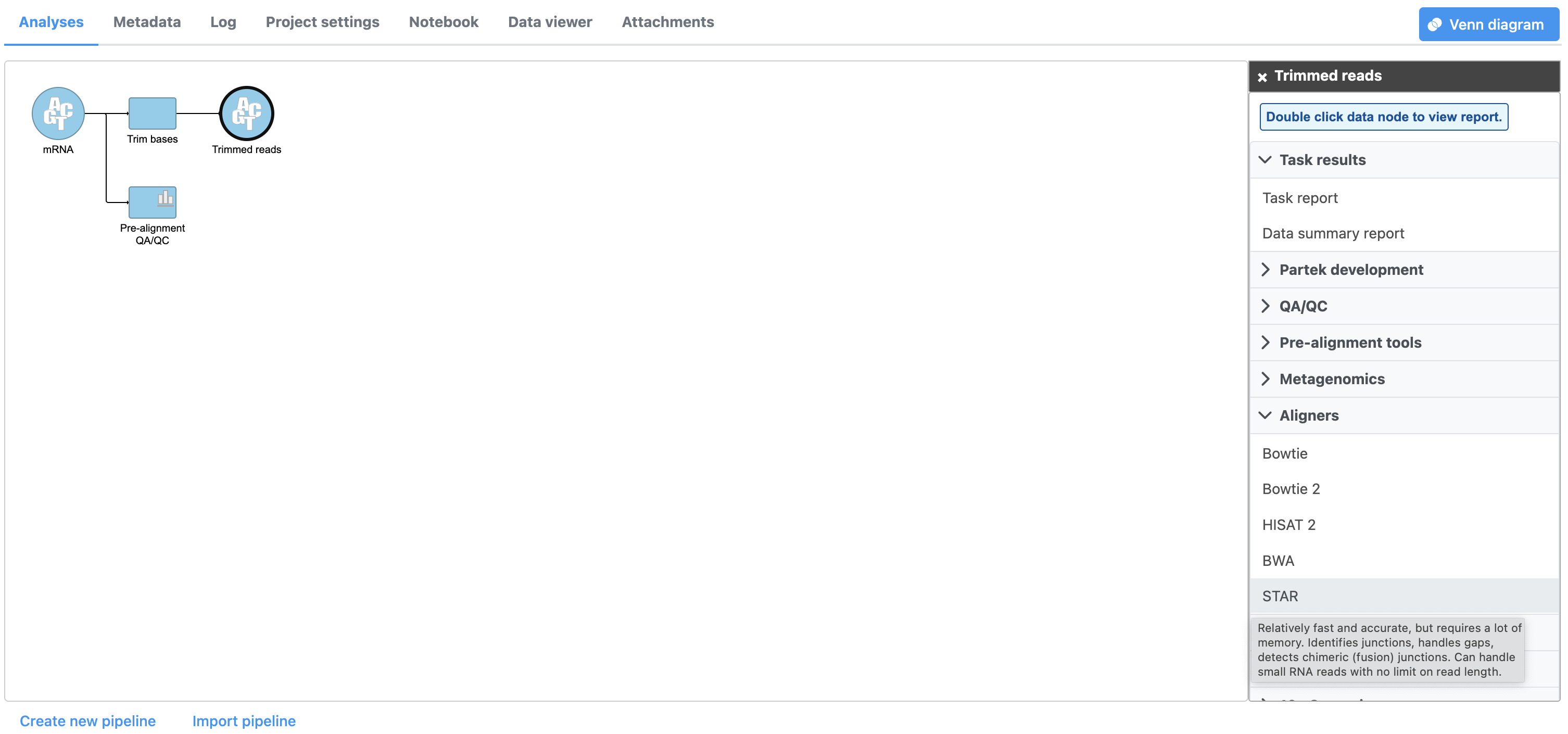
Figure 1. A variety of different aligners are available in Partek Flow. In this tutorial we will use STAR aligner, a popular choice for aligning RNA-Seq data.
Partek Flow offers a variety of different aligners. Mouse over any option for a short description. For this tutorial, we will use STAR, a fast and accurate aligner commonly used for RNA-Seq data. For more information about STAR and the other aligners, please consult the Aligners user guide.
- Click STAR
The STAR aligner options allow us to select the genome build (assembly) and index. For this tutorial, our data set contains only reads that map to chromosome 22 to minimize the time required for resource-intensive tasks, such as alignment.
- Click Finish to run with hg19 selected for Assembly and Whole genome for the Aligner index (Figure 2)
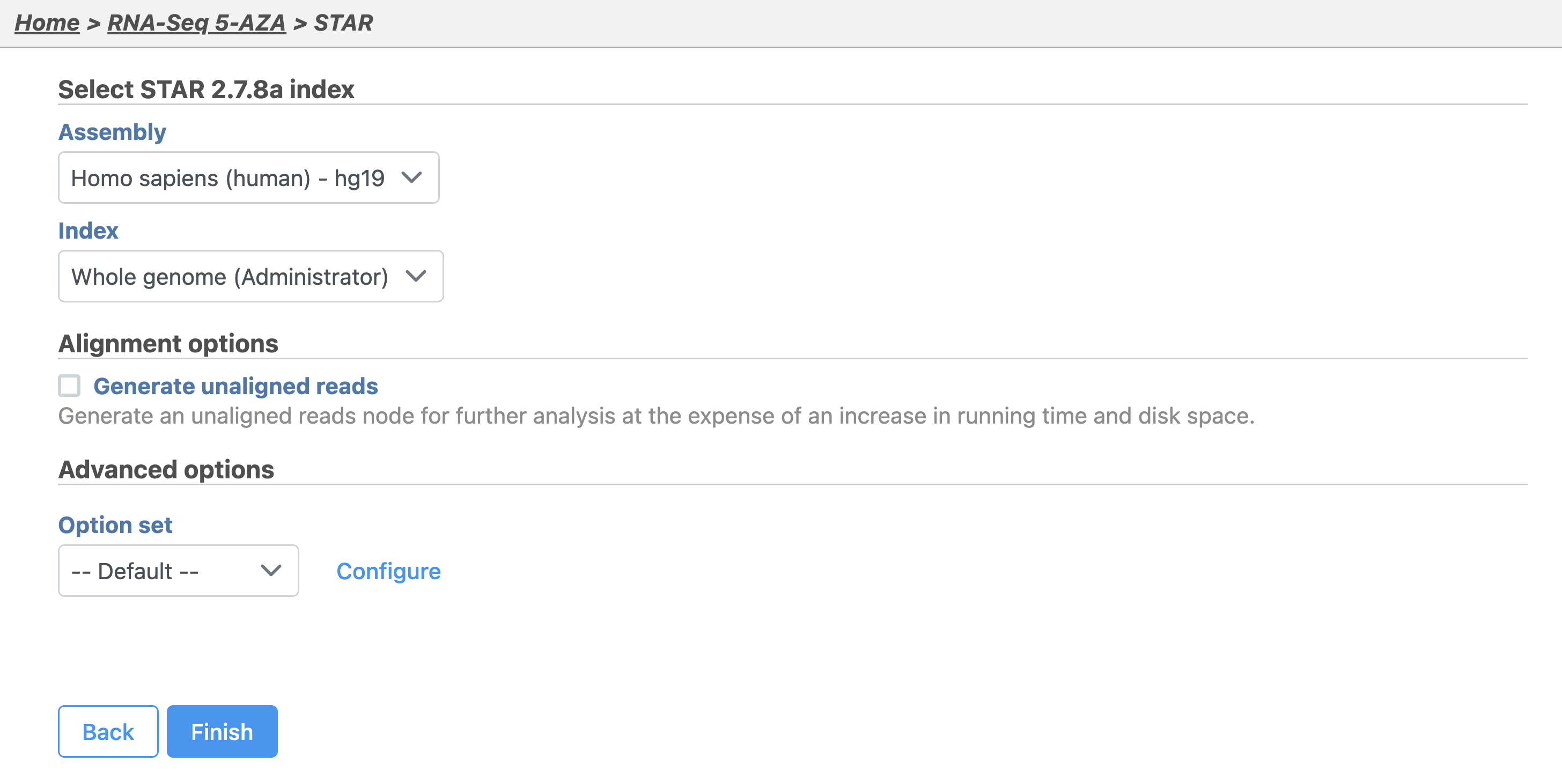
Figure 2. Configuring the STAR aligner
Alignment is a resource-intensive task and may take around 20 minutes to complete, even when mapping only reads from a single chromosome. Task and data nodes that have been queued, but not completed, are shown in a ligher color than completed tasks (Figure 3).
Figure 3. Queued tasks and their output data nodes are shown in a lighter color
The Align reads task generates an Aligned reads data node once complete. You can wait for the alignment task to finish or you can continue building the pipeline while the results of alignment are pending; additional tasks can be added to the pipeline and queued before the current task has completed.
Additional Assistance
If you need additional assistance, please visit our support page to submit a help ticket or find phone numbers for regional support.


| Your Rating: |
|
Results: |

|
30 | rates |
Overview
Content Tools