Based on pre-alignment QA/QC, we need to trim low quality bases from the 3' end of reads.
- Click the Unaligned reads data node
- Click Pre-alignment tools in the task menu
- Click Trim bases (Figure 1)
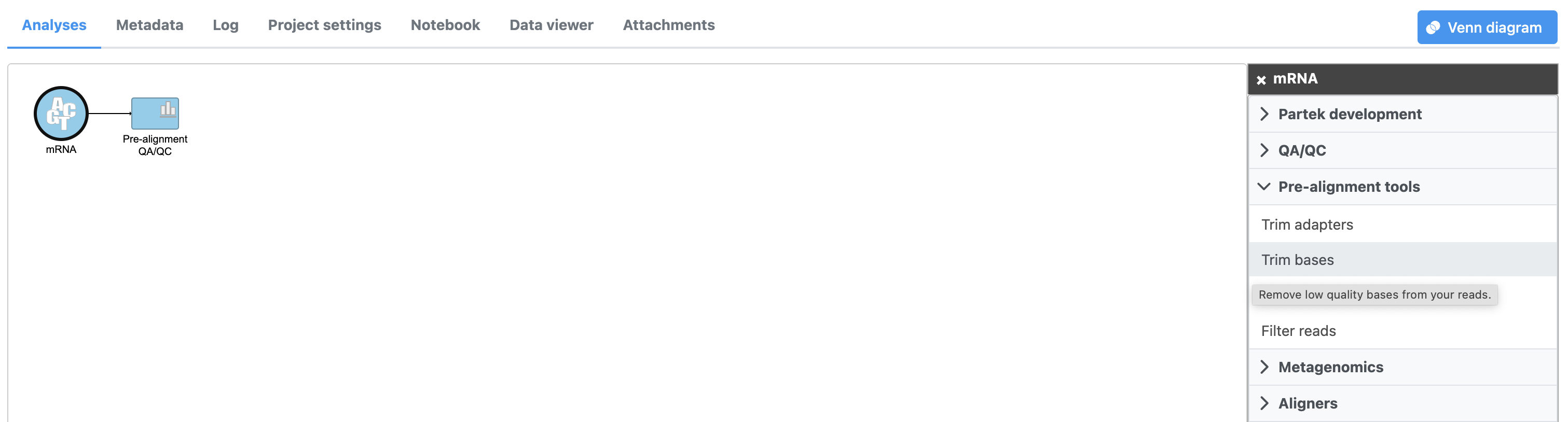
Figure 1. Invoking the Trim bases task
By default, Trim bases removes bases starting at the 3' end and continuing until it finds a base pair call with a Phred score of equal to or greater than 35 (Figure 2).
- Click Finish to run Trim bases with default settings
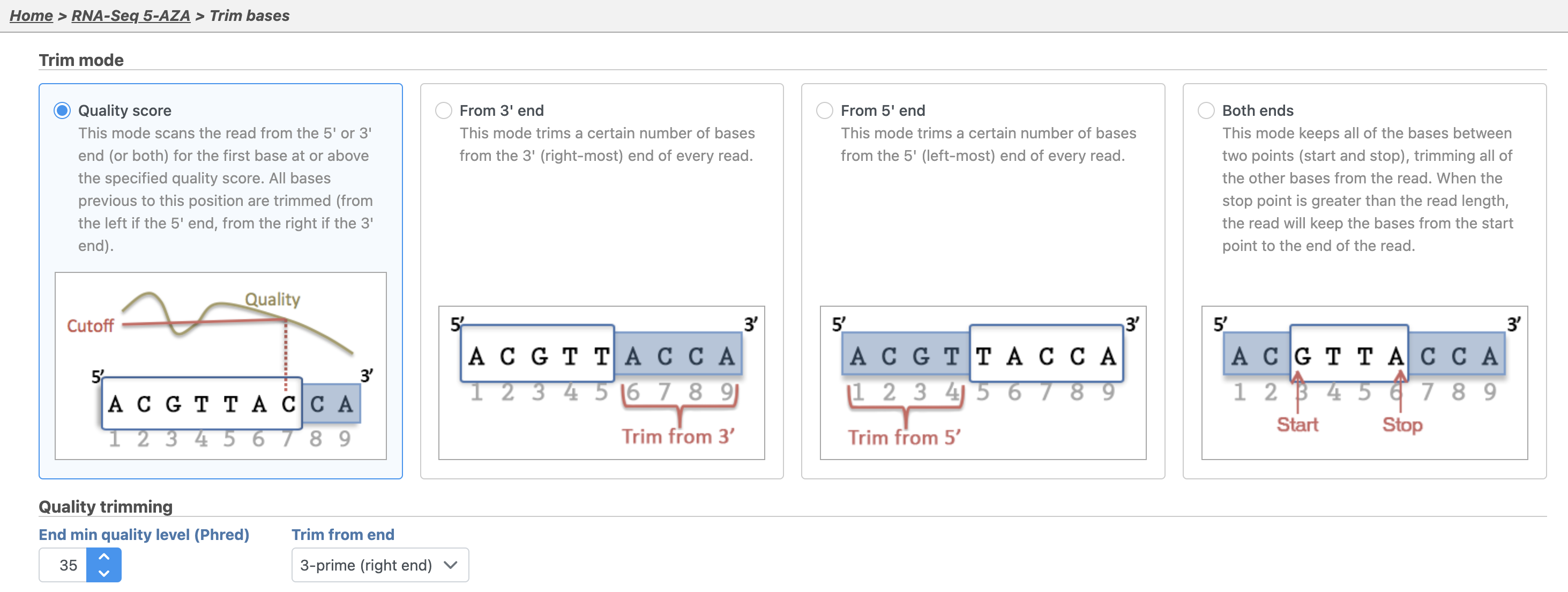
Figure 2. Configuring Trim bases
The Trim bases task will generate a new data node, Trimmed reads (Figure 3). We can view the task report for Trim bases by double-clicking either the Trim bases task node or the Trimmed reads data node or choosing Task report from the task menu.
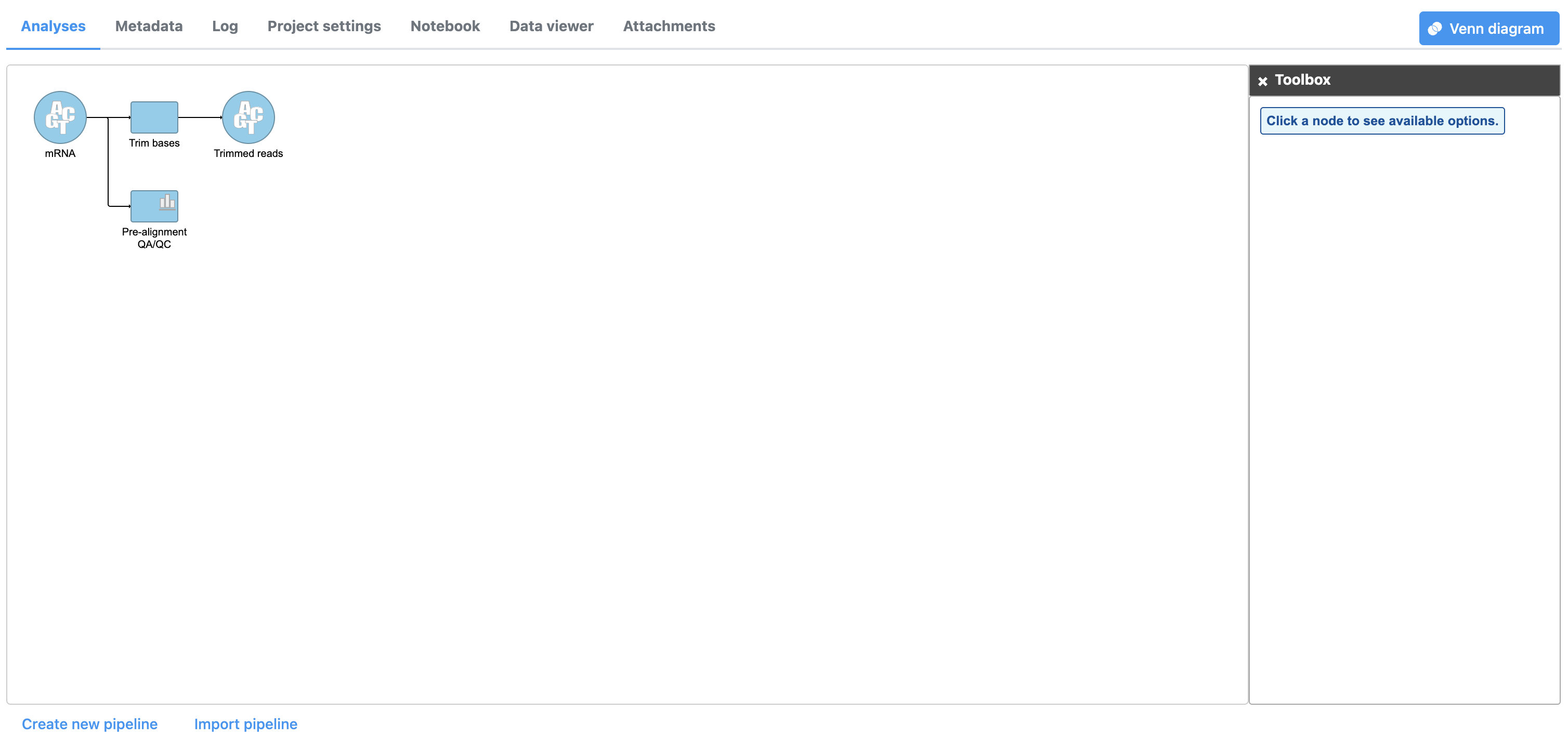 Figure 3. A Task and a Data node are created from the Trim bases task. Task and Data nodes that have been queued or are in progress are shown in a lighter color than completed tasks.
Figure 3. A Task and a Data node are created from the Trim bases task. Task and Data nodes that have been queued or are in progress are shown in a lighter color than completed tasks.
- Double-click the Trimmed reads data node to open the task report
The report shows the percentage of trimmed reads and reads removed in a spreadsheet and a two graphs (Figure 4).
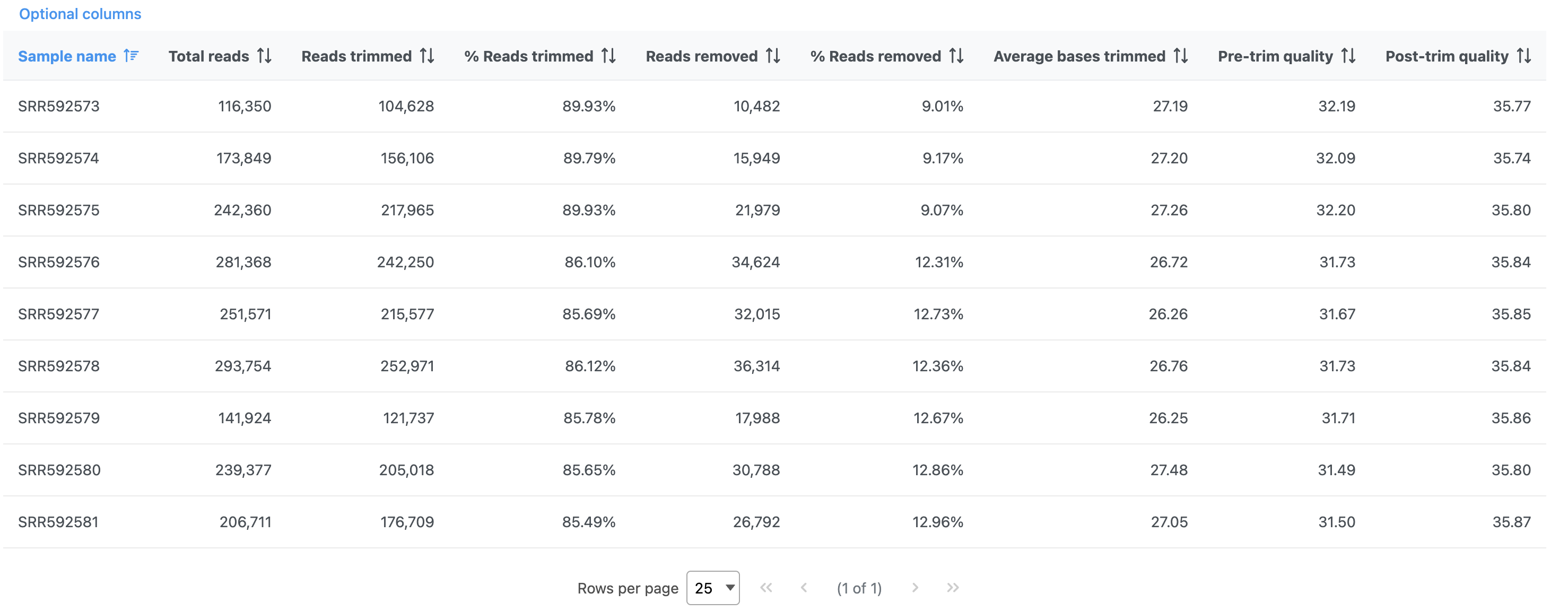
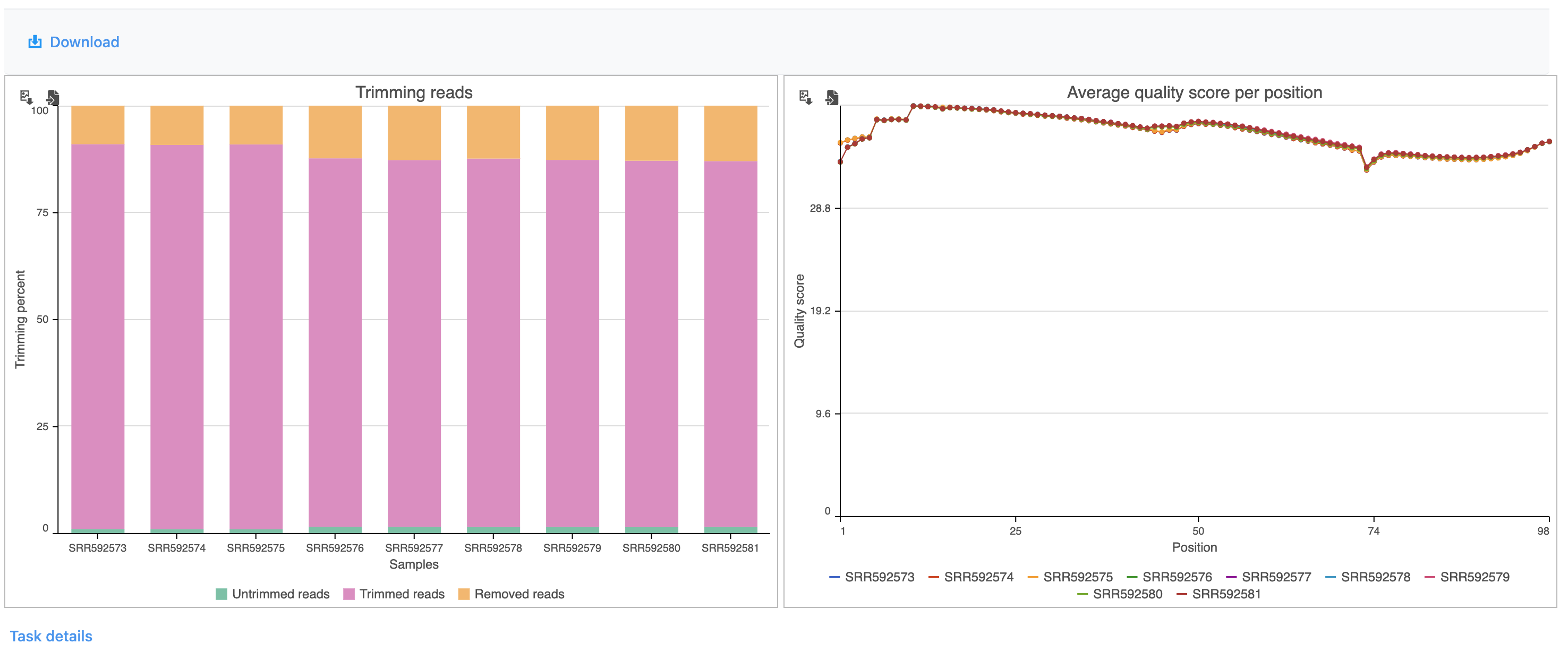
Figure 4. Results of the Trim bases task
The results are fairly consistent across samples with ~2% of reads untrimmed, ~86% trimmed, and ~12% removed for each. The average quality score for each sample is increased with higher average quality scores at the 3' ends.
- Click RNA-Seq 5-AZA to return to the Analyses tab
Additional Assistance
If you need additional assistance, please visit our support page to submit a help ticket or find phone numbers for regional support.


| Your Rating: |
|
Results: |

|
30 | rates |
Overview
Content Tools