Split matrix
The Single cell counts data node contains two different types of data, mRNA expression and protein expression. So that we can process these two different types of data separately, we will split the data by data type.
- Click the Single cell counts data node
- Click Pre-analysis tools in the toolbox
- Click Split by feature type
A rectangular task node will be created along with two circular data nodes, one for each data type (Figure 1). The labels for these data types are determined by features.csv file used when processing the data with Cell Ranger. Here, our data is labeled Gene Expression, for the mRNA data, and Antibody Capture, for the protein data.
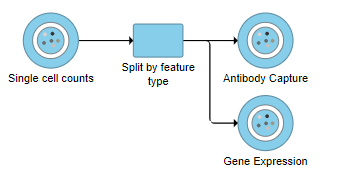 Figure 1. Split by feature type produces two data nodes, one for each data type
Figure 1. Split by feature type produces two data nodes, one for each data type
Filter low-quality cells
An important step in analyzing single cell RNA-Seq data is to filter out low-quality cells. A few examples of low-quality cells are doublets, cells damaged during cell isolation, or cells with too few counts to be analyzed. In a CITE-Seq experiment, protein aggregation in the antibody staining reagents can cause a cell to have a very high number of counts. These are low-quality cells that can be excluded. Additionally, if all cells in a data set are expected to show a baseline level of expression for one of the antibodies used, it may be appropriate to filter out cells with very low counts or a low number of detected features. You can do this in Partek Flow using the Single cell QA/QC task.
We will start with the protein data.
- Click the Antibody Capture data node
- Click QA/QC in the toolbox
- Click Single Cell QA/QC
This produces a Single-cell QA/QC task node (Figure 2).
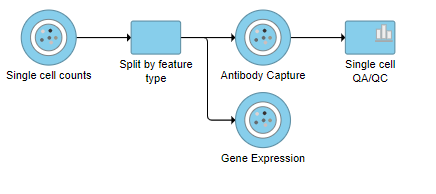 Figure 2. Single cell QA/QC produces a task node
Figure 2. Single cell QA/QC produces a task node
- Double-click the Single cell QA/QC task node to open the task report
The Single cell QA/QC report opens in a new data viewer session. There are interactive violin plots showing the most commonly used quality metrics for each cell: the total count per cell and the number of detected features per cell (Figure 3). Each point on the plots is a cell and the violins illustrate the distribution of values for the y-axis metric.
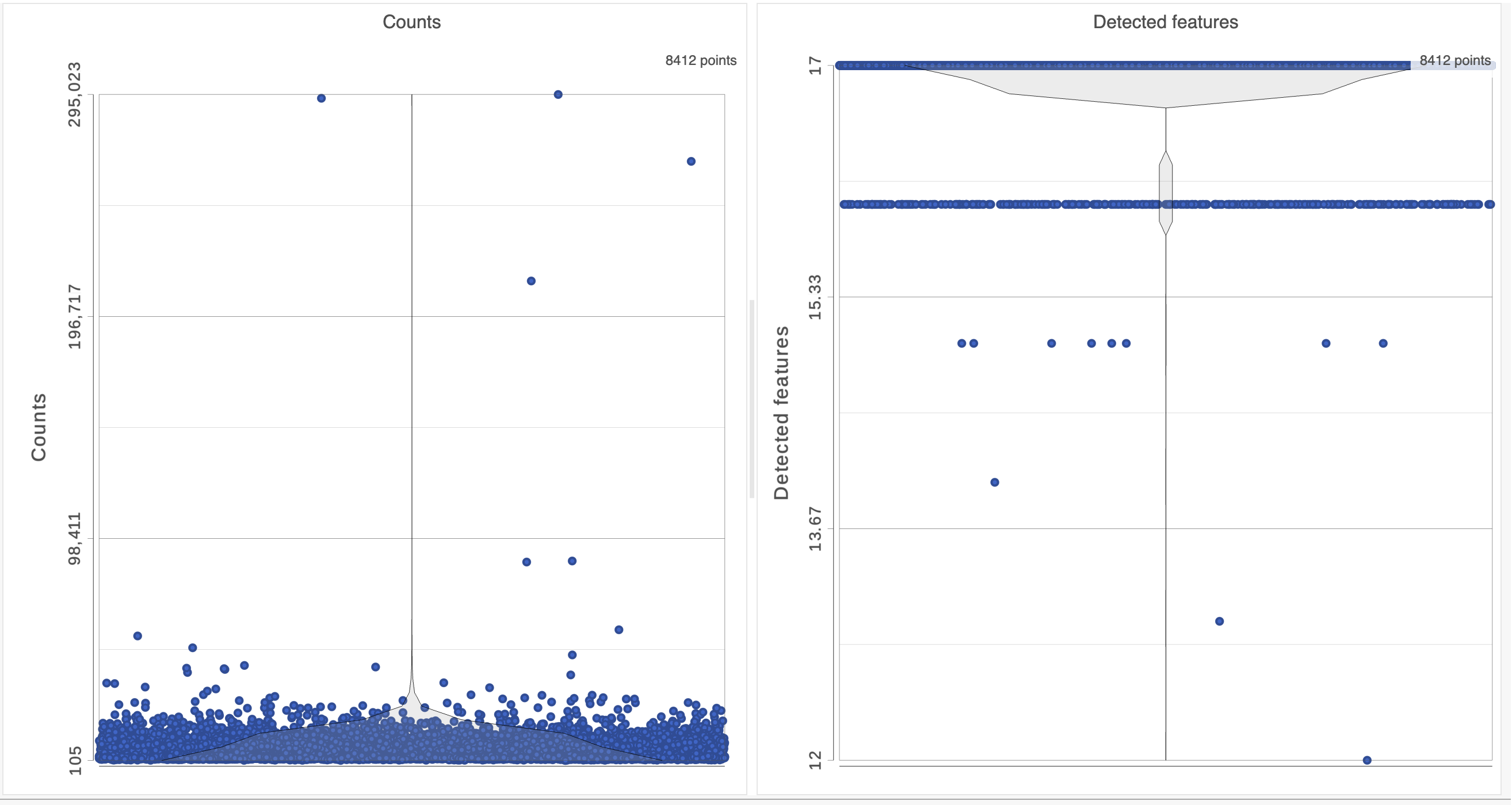 Figure 3. Each cell is shown as a point on the plot.
For this analysis, we will set a maximum counts threshold to exclude potential protein aggregates and, because we expect every cell to be bound by several antibodies, we will also set a minimum counts threshold.
Figure 3. Each cell is shown as a point on the plot.
For this analysis, we will set a maximum counts threshold to exclude potential protein aggregates and, because we expect every cell to be bound by several antibodies, we will also set a minimum counts threshold.
- Select one of the plots on the canvas
- In the Select & Filter icon on the left under Tools, set the Counts threshold to keep cells between 500 and 20000 (Figure 4)
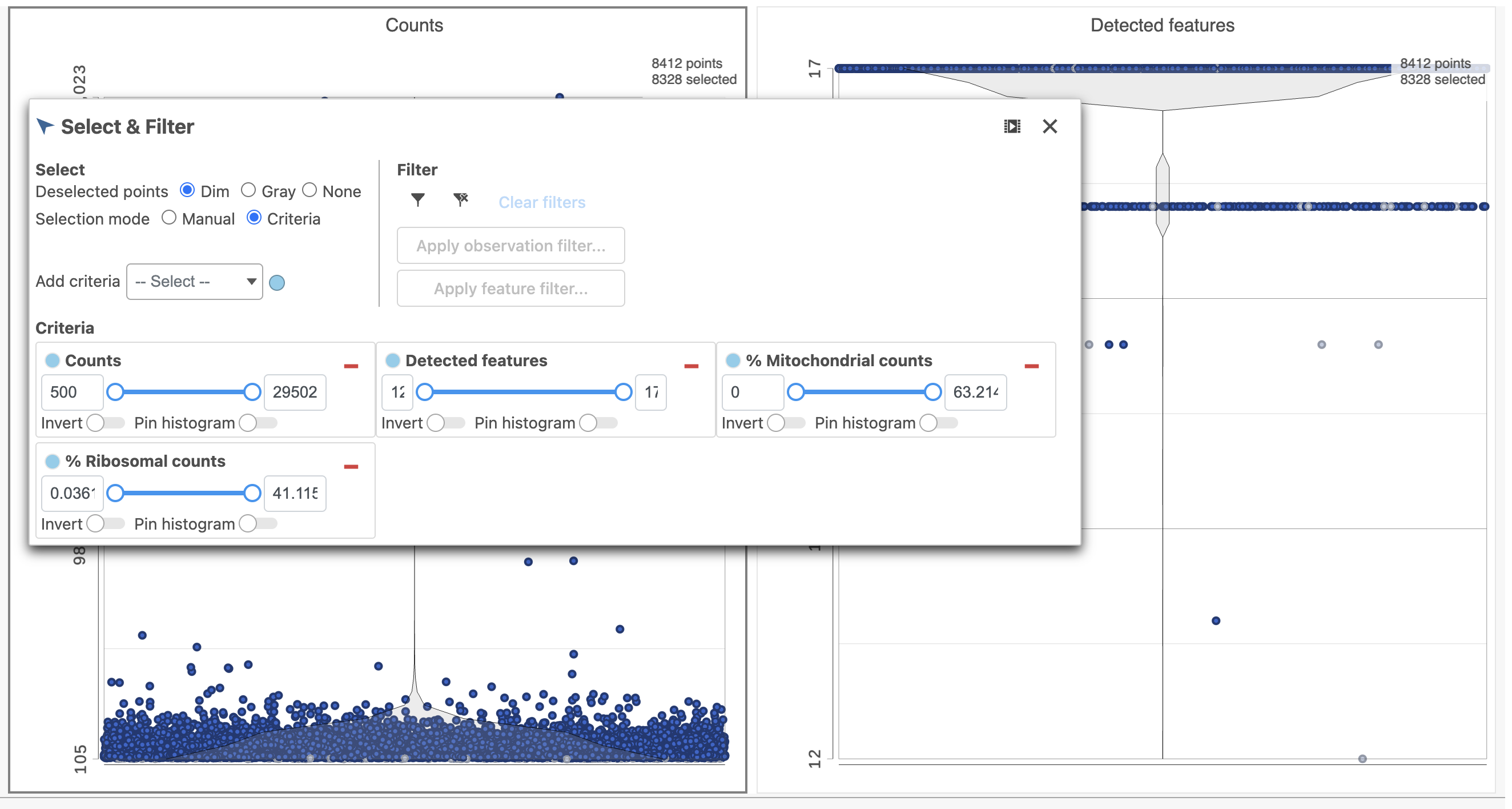 Figure 4. Filtering low quality cells based on protein expression data
Figure 4. Filtering low quality cells based on protein expression data
- Click
 under Filter on the right
under Filter on the right - Click Apply observation filter...
- Select the Antibody Capture data node as input in the pipeline preview (Figure 5)
- Click Select
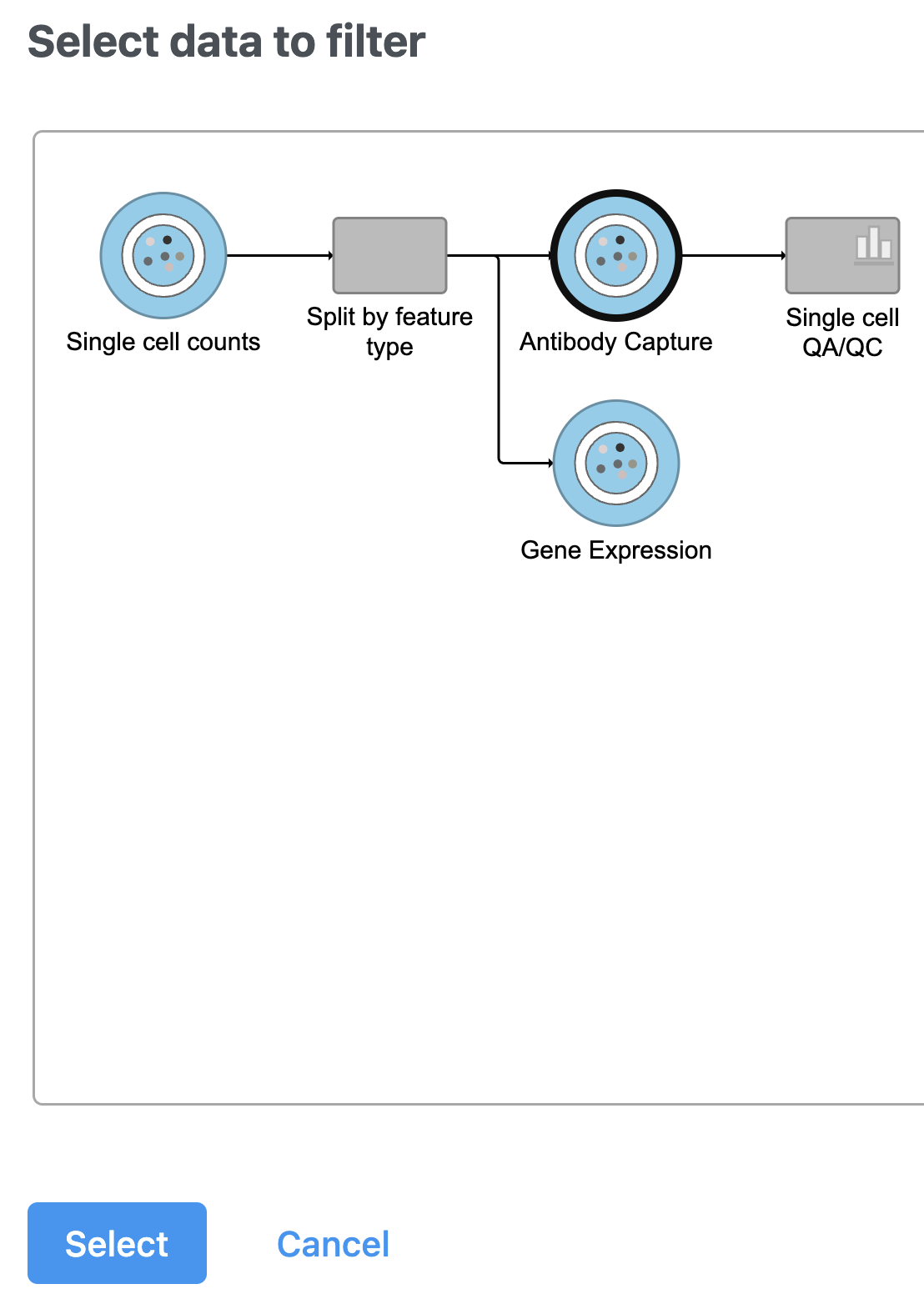 Figure 5. After the Apply filter button is selected, you will be presented with a preview of your pipeline. You need to select the appropriate data node to apply the filtering to. In this case, the Antibody capture node
You will see a message telling you a new task has been enqueued.
Figure 5. After the Apply filter button is selected, you will be presented with a preview of your pipeline. You need to select the appropriate data node to apply the filtering to. In this case, the Antibody capture node
You will see a message telling you a new task has been enqueued.
- Click OK to dismiss the message
- Click the project name at the top to go back to the Analyses tab
- Your browser may warn you that any unsaved changes to the data viewer session will be lost. Ignore this message and proceed to the Analyses tab
A new task, Filter counts, is added to the Analyses tab. This task produces a new Filter counts data node.
Next, we can repeat this process for the Gene Expression data node.
- Click the Gene Expression data node
- Click the QA/QC section in the toolbox
- Click Single Cell QA/QC
This produces a Single-cell QA/QC task node
- Double-click the Single cell QA/QC task node to open the task report
The task report lists the number of counts per cell, the number of detected features per cell, the percentage of mitochondrial reads per cell, and the percentage of ribosomal counts per cell in four violin plots (Figure 6). For this analysis, we will set maximum and minimum thresholds for total counts and detected genes to exclude potential doublets and a maximum mitochondrial reads percentage filter to exclude potential dead or dying cells. There is no need to apply a filter based on the percentage of ribosomal counts in this tutorial.
- In the Selection card on the right, set the Counts threshold to keep cells between 1500 and 15000
- Set the Detected features to keep cells between 400 and 4000
- Set the % Mitochondrial counts to keep cells between 0% and 20% (Figure 6)
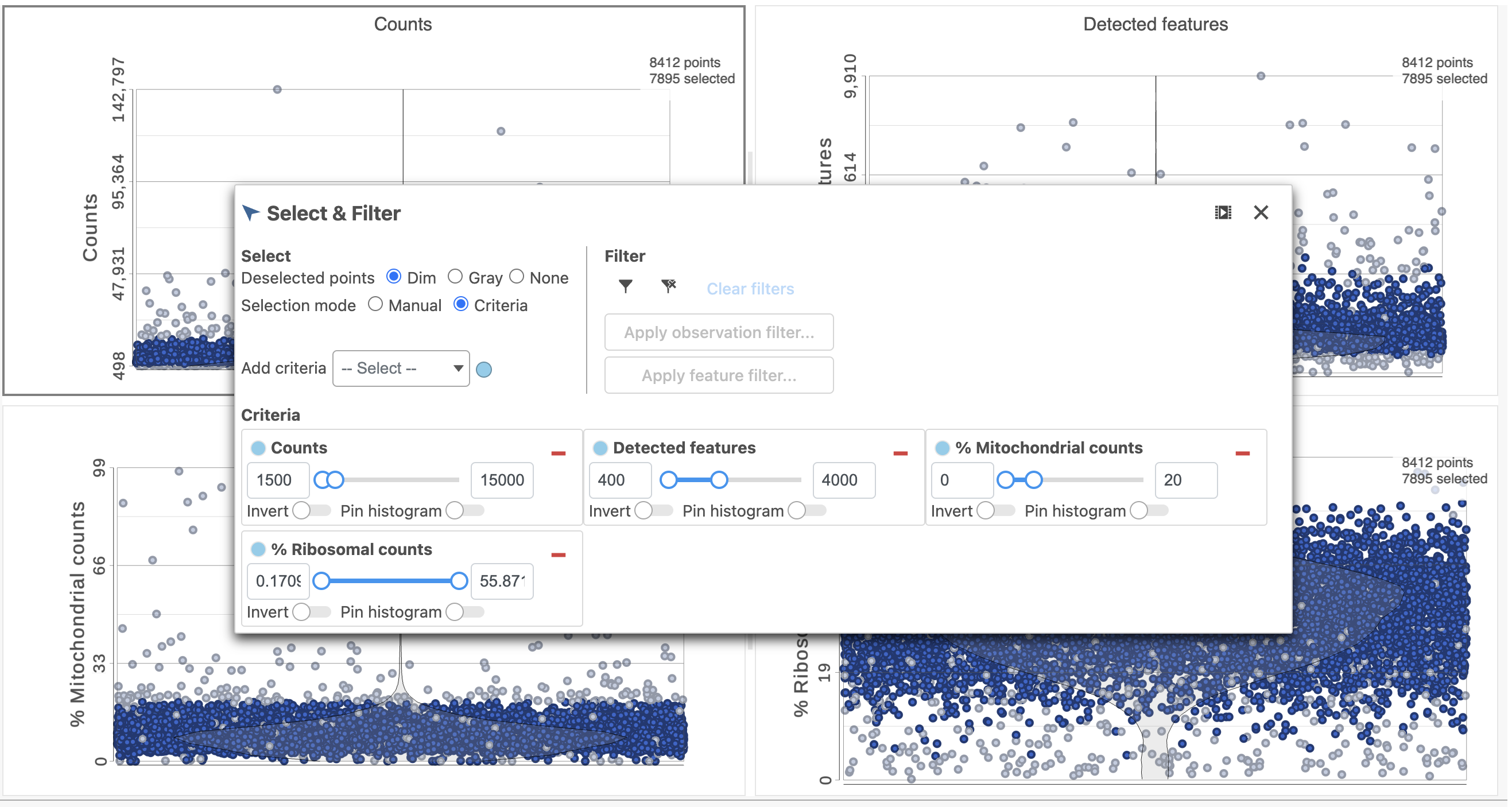 Figure 6. Filtering low quality cells based on gene expression data
Figure 6. Filtering low quality cells based on gene expression data
- Click under Filter on the right
- Click Apply observations filter
- Select the Gene Expression data node as input in the pipeline preview
- Click Select
- Click OK to dismiss the message about the task being enqueued
- Click the project name at the top to go back to the Analyses tab
- Your browser may warn you that any unsaved changes to the data viewer session will be lost. Ignore this message and proceed to the Analyses tab
A new task, Filter counts, is added to the Analyses tab. This task produces a new Filter counts data node (Figure 7)
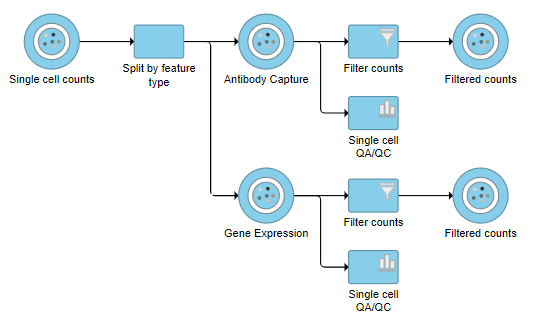 Figure 7. Antibody Capture and Gene Expression data have been filtered to remove low quality cells
Figure 7. Antibody Capture and Gene Expression data have been filtered to remove low quality cells
Normalization
After excluding low-quality cells, we can normalize the data.
We will start with the protein data.
- Click the Filtered counts data node produced by filtering the Antibody Capture data node
- Click Normalization and scaling in the toolbox
- Click Normalization
- Click the green
 button
button - Click Finish to run (Figure 8)
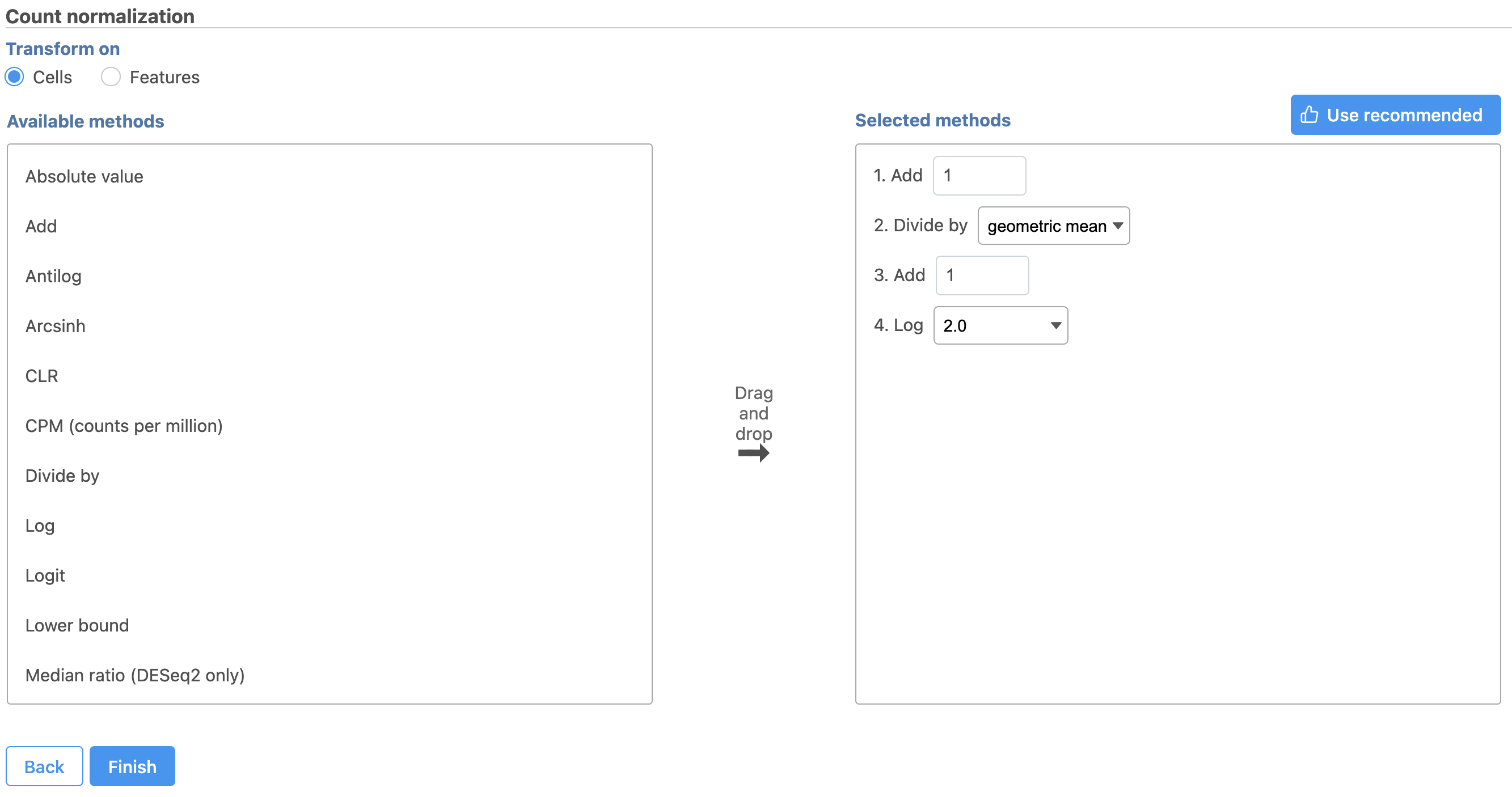 Figure 8. Recommended normalization for protein count data
The recommended normalization for protein data includes the following steps: Add 1, Divide by Geometric mean, Add 1, Log base 2. This is a variant of Centered log-ratio (CLR), which was used to normalize antibody capture protein counts data in the paper that introduced CITE-Seq [1] and in subsequent publications on similar assays [2. 3]. CLR normalization includes the following steps: Add 1, Divide by Geometric mean, Add 1, log base e. Normalizing the protein data to base 2 instead of e allows for better integration with gene expression data further downstream. If you would prefer to use CLR, click and drag CLR from the panel on the left to the right. If you do choose to use CLR, we recommend making sure the gene expression data is normalized to the base e, to allow for smoother integration further downstream.
Figure 8. Recommended normalization for protein count data
The recommended normalization for protein data includes the following steps: Add 1, Divide by Geometric mean, Add 1, Log base 2. This is a variant of Centered log-ratio (CLR), which was used to normalize antibody capture protein counts data in the paper that introduced CITE-Seq [1] and in subsequent publications on similar assays [2. 3]. CLR normalization includes the following steps: Add 1, Divide by Geometric mean, Add 1, log base e. Normalizing the protein data to base 2 instead of e allows for better integration with gene expression data further downstream. If you would prefer to use CLR, click and drag CLR from the panel on the left to the right. If you do choose to use CLR, we recommend making sure the gene expression data is normalized to the base e, to allow for smoother integration further downstream.
Normalization produces a Normalized counts data node on the Antibody Capture branch of the pipeline.
Next, we can normalize the mRNA data. We will use the recommended normalization method in Partek Flow, which accounts for differences in library size, or the total number of UMI counts, per cell, adds 1 and log2 transforms the data.
- Click the Filtered counts data node produced by filtering the Gene Expression data node
- Click the Normalization and scaling section in the toolbox
- Click Normalization
- Click the button
- Click Finish to run (Figure 9)
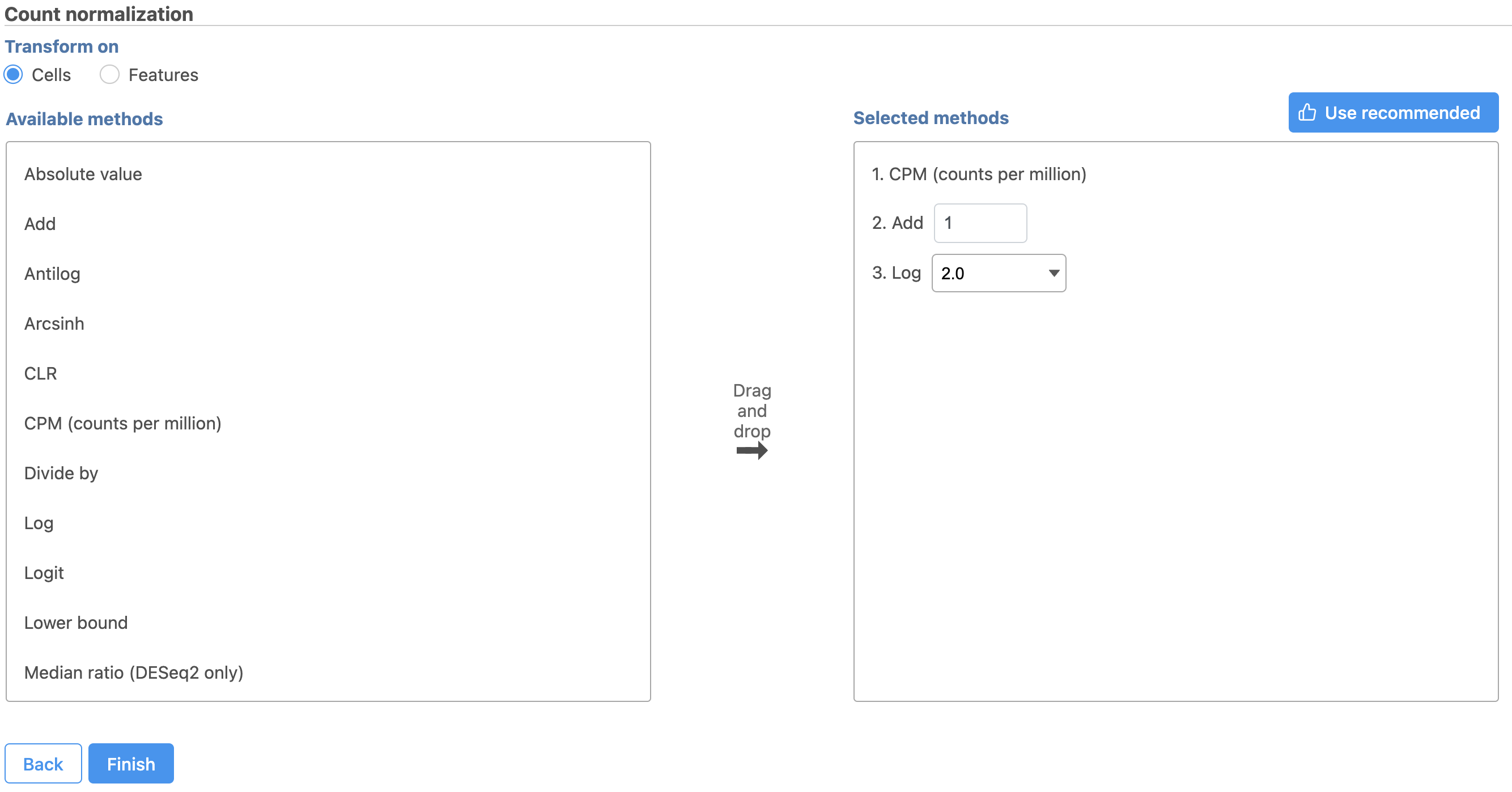 Figure 9. Recommended normalization for single cell gene expression data
Normalization produces a Normalized counts data node on the Gene Expression branch of the pipeline (Figure 10).
Figure 9. Recommended normalization for single cell gene expression data
Normalization produces a Normalized counts data node on the Gene Expression branch of the pipeline (Figure 10).
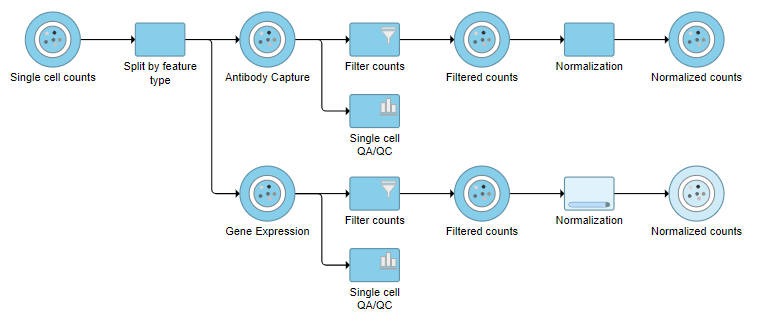 Figure 10. The two normalization tasks produce Normalized counts data nodes
Figure 10. The two normalization tasks produce Normalized counts data nodes
Merge Protein and mRNA data
For quality filtering and normalization, we needed to have the two data types separate as the processing steps were distinct. For downstream analysis, we want to be able to analyze protein and mRNA data together. To bring the two data types back together, we will merge the two normalized counts data nodes.
- Click the Normalized counts data node on the Antibody Capture branch of the pipeline
- Click Pre-analysis tools in the toolbox
- Click Merge matrices
- Click Select data node to launch the data node selector
Data nodes that can be merged with the Antibody Capture branch Normalized counts data node are shown in color (Figure 11).
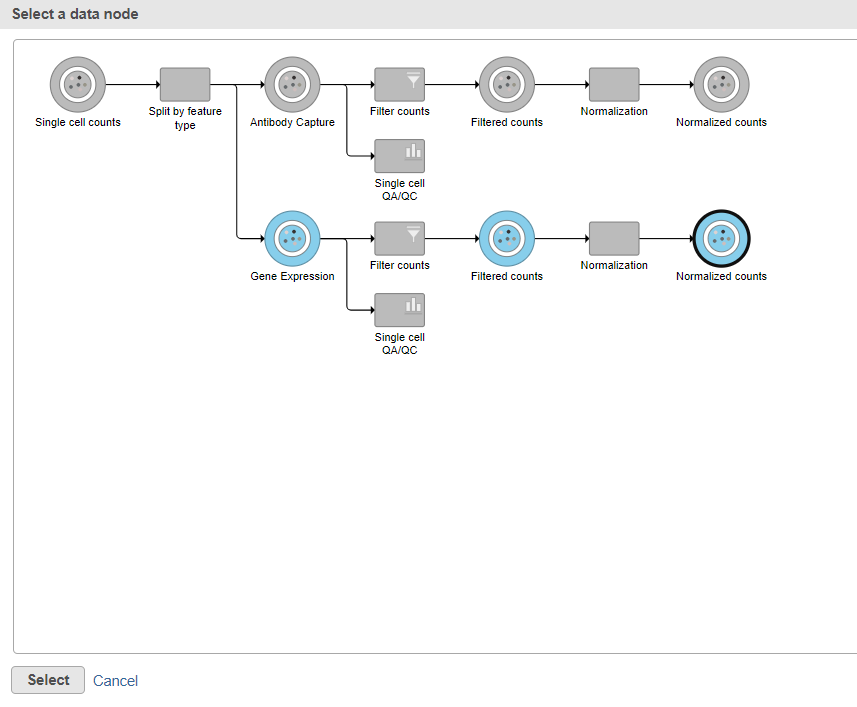 Figure 11. Select the normalizated gene expression counts to merge the protein counts with
Figure 11. Select the normalizated gene expression counts to merge the protein counts with
- Click the Normalized counts data node on the Gene Expression branch of the pipeline (Figure 11)
- Click Select
- Click Finish to run the task
The output is a Merged counts data node (Figure 12). This data node will include the normalized counts of our protein and mRNA data. The intersection of cells from the two input data nodes is retained so only cells that passed the quality filter for both protein and mRNA data will be included in the Merged counts data node.
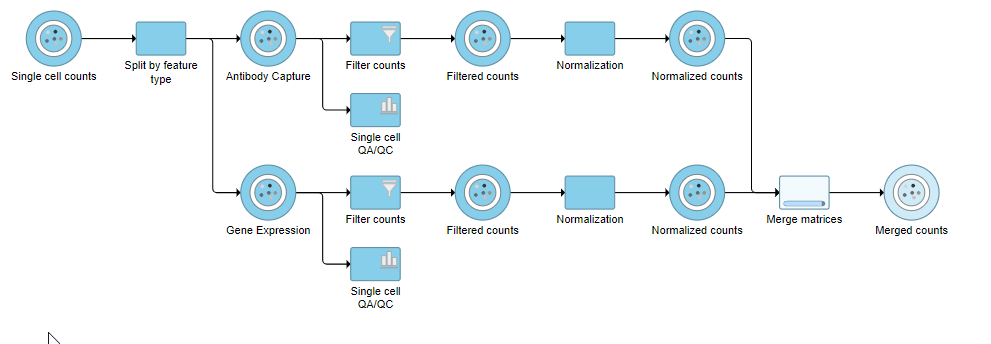 Figure 12. Merged counts output
Figure 12. Merged counts output
Collapsing tasks to simplify the pipeline
To simplify the appearance of the pipeline, we can group task nodes into a single collapsed task. Here, we will collapse the filtering and normalization steps.
- Right-click the Split by feature type task node
- Choose Collapse tasks from the pop-up dialog (Figure 13)
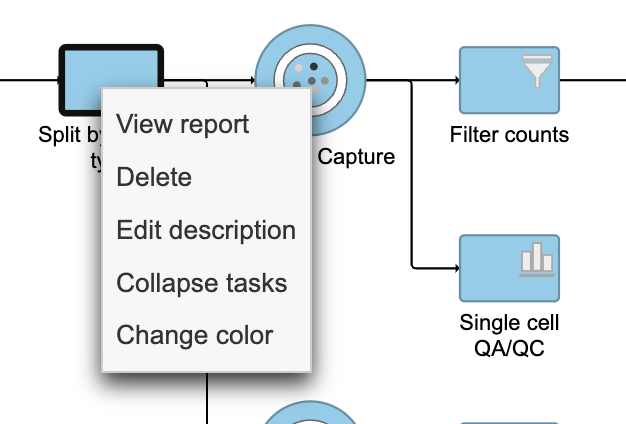 Figure 13. Choosing the first task node to generate a collapsed task
Tasks that can be selected for the beginning and end of the collapsed section of the pipeline are highlighted in purple (Figure 14). We have chosen the Split matrix task as the start and we can choose Merge matrices as the end of the collapsed section.
Figure 13. Choosing the first task node to generate a collapsed task
Tasks that can be selected for the beginning and end of the collapsed section of the pipeline are highlighted in purple (Figure 14). We have chosen the Split matrix task as the start and we can choose Merge matrices as the end of the collapsed section.
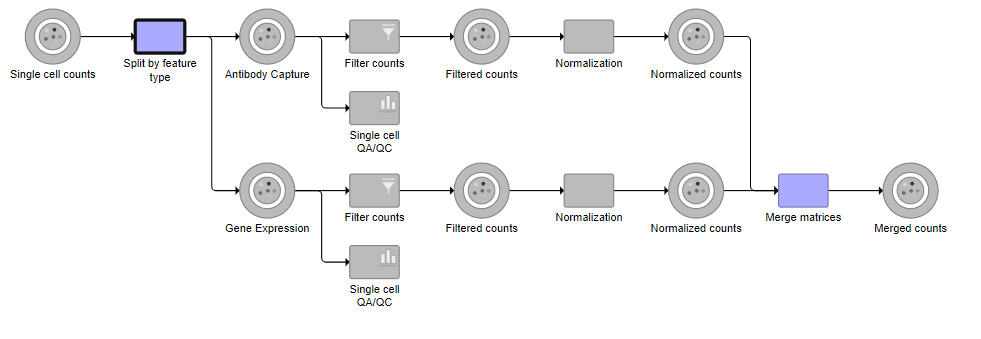 Figure 14. Tasks that can be the start or end of a collapsed task are shown in purple
Figure 14. Tasks that can be the start or end of a collapsed task are shown in purple
- Click the Merge matrices task to choose it as the end of the collapsed section
- Name the Collapsed task Data processing
- Click Save (Figure 15)
 Figure 15. Naming the collapsed task
Figure 15. Naming the collapsed task
The new collapsed task, Data processing, appears as a single rectangular task node (Figure 16).
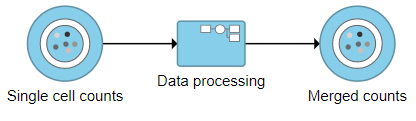 Figure 16. Collapsed tasks are represented by a single task node
To view the tasks in Data processing, we can expand the collapsed task.
Figure 16. Collapsed tasks are represented by a single task node
To view the tasks in Data processing, we can expand the collapsed task.
- Double-click Data processing to expand it or right-click and choose Expand collapsed task
When expanded, the collapsed task is shown as a shaded section of the pipeline with a title bar (Figure 17).
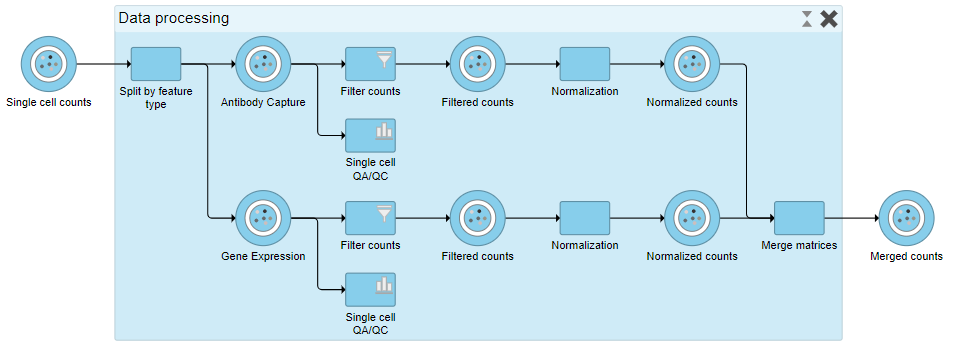 Figure 17. Expanding a collapsed task to show its components
To re-collapse the task, you can double click the title bar or click the
Figure 17. Expanding a collapsed task to show its components
To re-collapse the task, you can double click the title bar or click the ![]() icon in the title bar. To remove the collapsed task, you can click the
icon in the title bar. To remove the collapsed task, you can click the ![]() . Please note that this will not remove tasks, just the grouping.
. Please note that this will not remove tasks, just the grouping.
- Double-click the Data processing title bar to re-collapse
References
[1] Stoeckius, M., Hafemeister, C., Stephenson, W., Houck-Loomis, B., Chattopadhyay, P. K., Swerdlow, H., ... & Smibert, P. (2017). Simultaneous epitope and transcriptome measurement in single cells. Nature methods, 14(9), 865.
[2] Stoeckius, M., Zheng, S., Houck-Loomis, B., Hao, S., Yeung, B. Z., Mauck, W. M., ... & Satija, R. (2018). Cell hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome biology, 19(1), 224.
[3] Mimitou, E., Cheng, A., Montalbano, A., Hao, S., Stoeckius, M., Legut, M., ... & Satija, R. (2018). Expanding the CITE-seq tool-kit: Detection of proteins, transcriptomes, clonotypes and CRISPR perturbations with multiplexing, in a single assay. bioRxiv, 466466.
Additional Assistance
If you need additional assistance, please visit our support page to submit a help ticket or find phone numbers for regional support.


| Your Rating: |
|
Results: |

|
10 | rates |
Overview
Content Tools