PCA
Next, we will perform some exploratory analysis on the merged mRNA and protein expression data and visualize the data in preparation to identify cell populations. Because the merged count matrix has thousands of features, it is a good idea to reduce the dimensionality of the data for more efficient downstream processing.
- Click the Merged counts data node
- Click Exploratory analysis in the toolbox
- Click PCA
- Click Finish to run the PCA with default settings (Figure 1)
 Figure 1. Run PCA with default settings
388pxA PCA task node will be added to the pipeline under the Analyses tab and a circular PCA output data node will be produced (Figure 2).
Figure 1. Run PCA with default settings
388pxA PCA task node will be added to the pipeline under the Analyses tab and a circular PCA output data node will be produced (Figure 2).
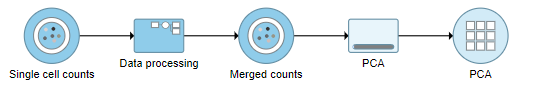 Figure 2. PCA task run on the merged counts data node
Once the task completes, we will inspect the results to decide the optimal number of principal components (PCs) to use in downstream analyses. To do this, we will use a Scree plot.
Figure 2. PCA task run on the merged counts data node
Once the task completes, we will inspect the results to decide the optimal number of principal components (PCs) to use in downstream analyses. To do this, we will use a Scree plot.
- Double click the PCA data node to open the task report
The PCA plot will open in a new data viewer session. A 3D scatterplot will be displayed on the canvas (Figure 3).
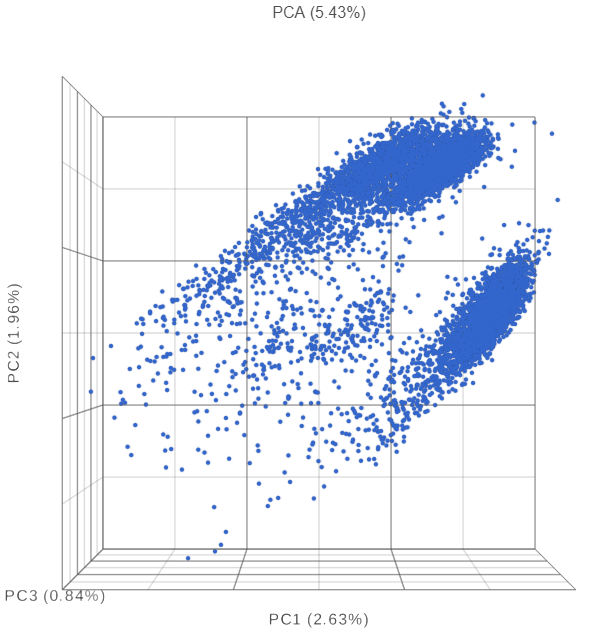 Figure 3. Each dot is a different cell. Cells are clustered based on how similar their expression profile is across the combined mRNA and protein data
Figure 3. Each dot is a different cell. Cells are clustered based on how similar their expression profile is across the combined mRNA and protein data
- Click and drag the Scree plot from New plot under Setup on the left onto the canvas
- Drop it over the Replace option (Figure 4)
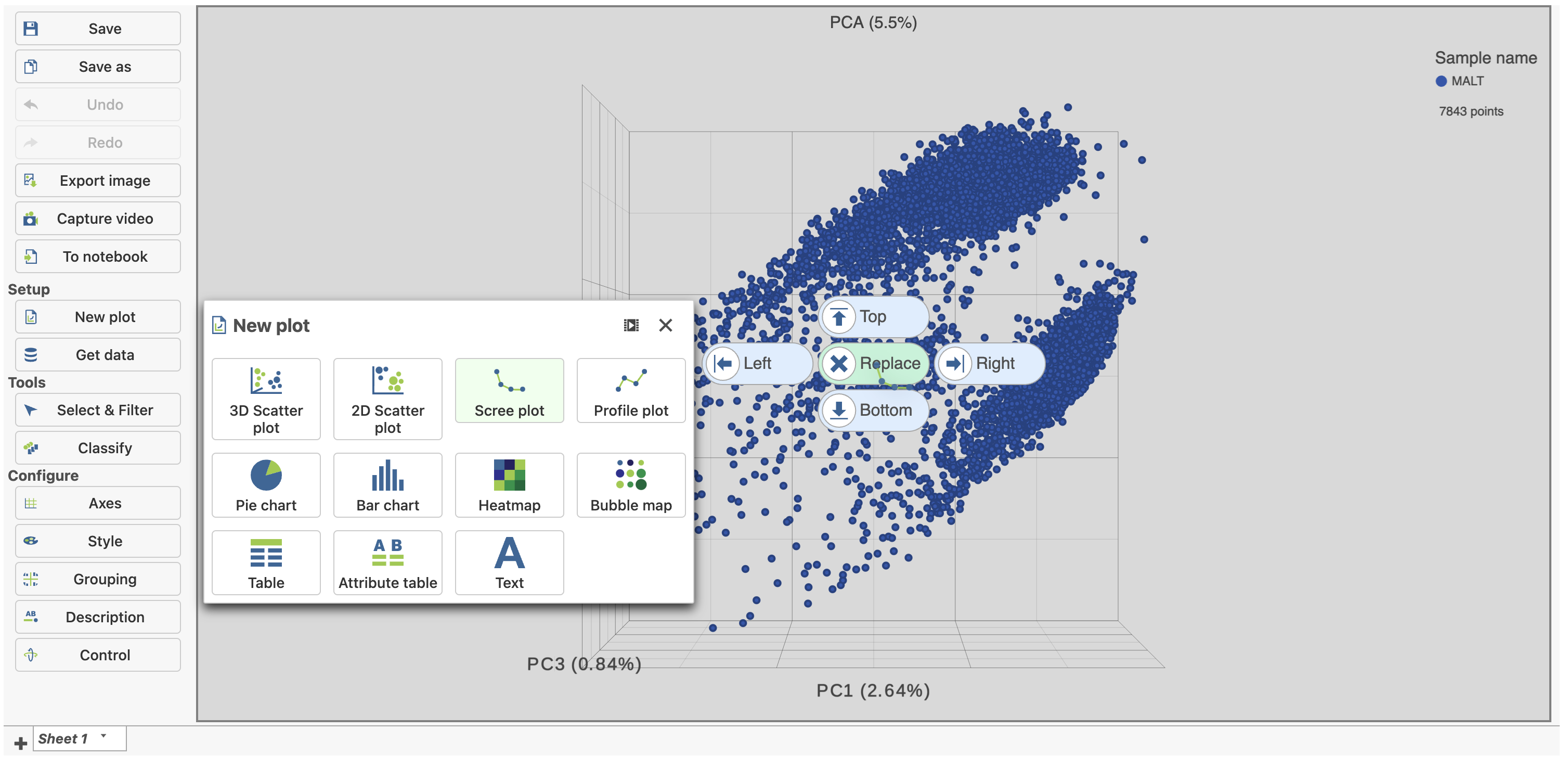 Figure 4. Click and drag the Scree plot to replace the PCA plot on the canvas
Figure 4. Click and drag the Scree plot to replace the PCA plot on the canvas
- Select PCA as data for the new Scree plot (Figure 5)
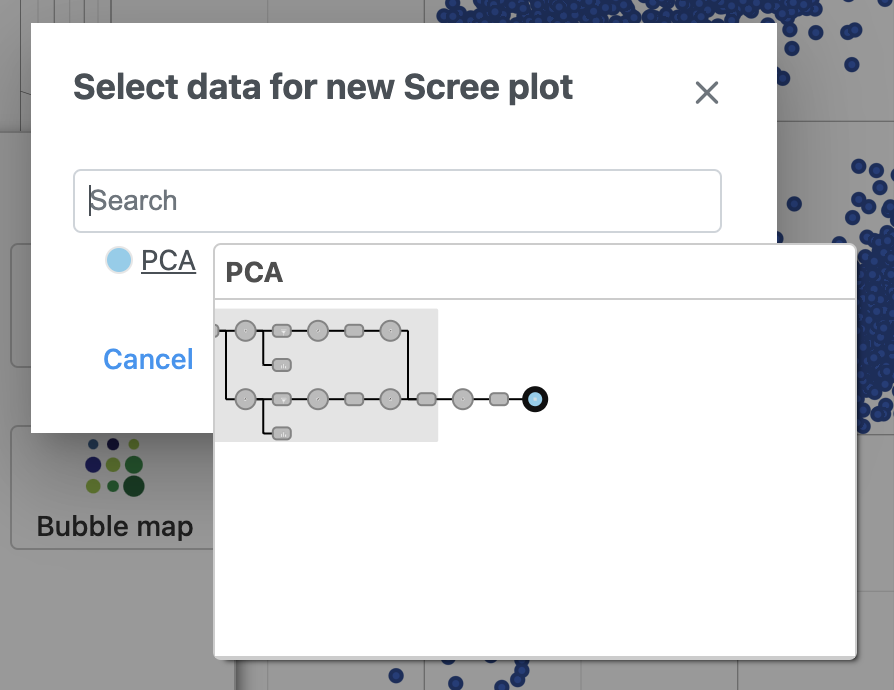 Figure 5. The PCA data node contains the data to draw the Scree plot
The Scree plot (Figure 6) shows the eigenvalues on the y-axis for each of the 100 PCs on the x-axis. The higher the eigenvalue, the more variance explained by each PC. Typically, after an initial set of highly informative PCs, the amount of variance explained by analyzing additional components is minimal. By identifying the point where the Scree plot levels off, you can choose an optimal number of PCs to use in downstream analysis steps like graph-based clustering and UMAP.
Figure 5. The PCA data node contains the data to draw the Scree plot
The Scree plot (Figure 6) shows the eigenvalues on the y-axis for each of the 100 PCs on the x-axis. The higher the eigenvalue, the more variance explained by each PC. Typically, after an initial set of highly informative PCs, the amount of variance explained by analyzing additional components is minimal. By identifying the point where the Scree plot levels off, you can choose an optimal number of PCs to use in downstream analysis steps like graph-based clustering and UMAP.
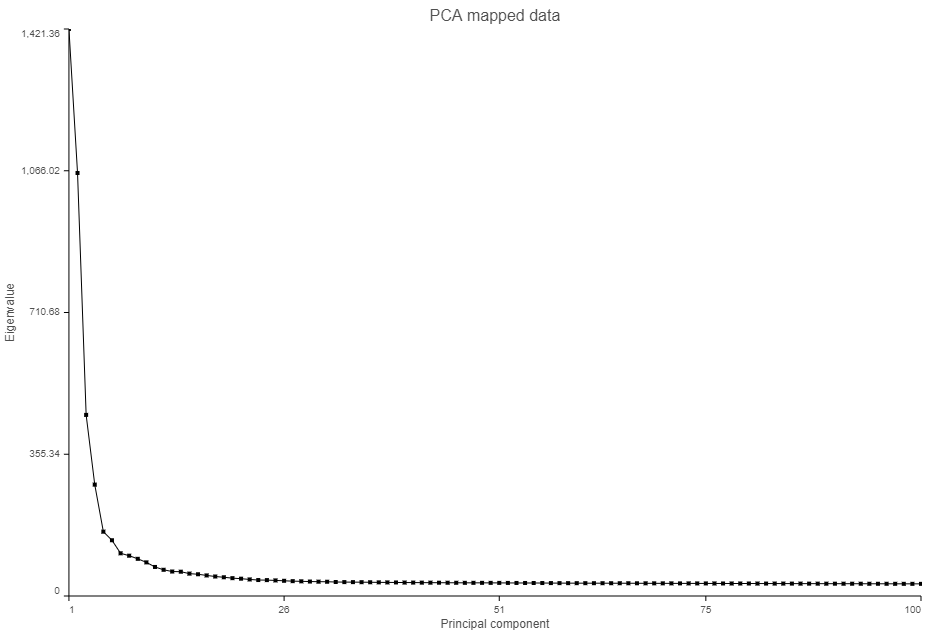 Figure 6. Scree plot shows the amount of variation explained by each principal component
Figure 6. Scree plot shows the amount of variation explained by each principal component
- Click and drag over the first set of PCs to zoom in (Figure 7)
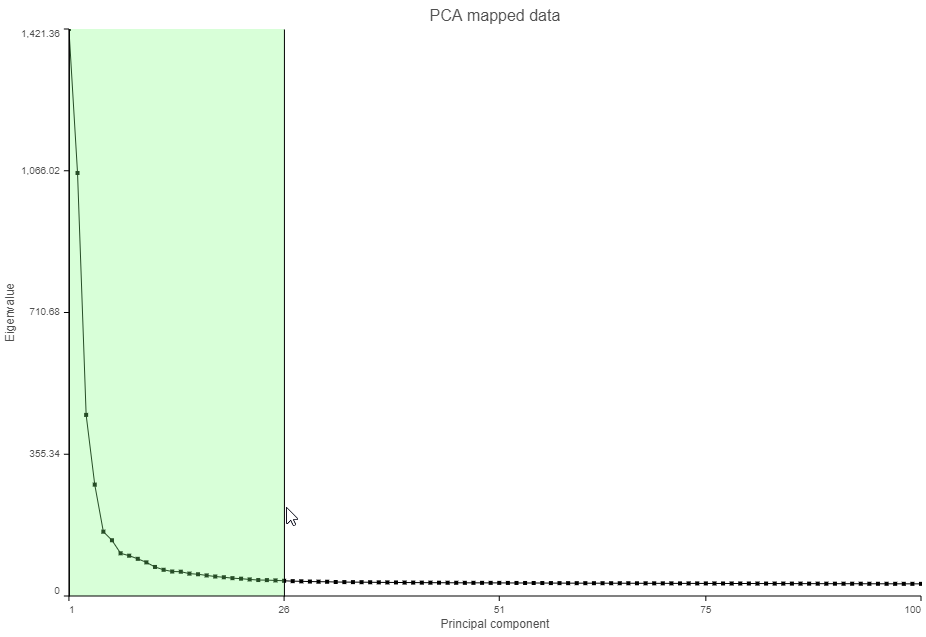 Figure 7. Click and drag on the Scree plot to zoom in and see the first set of principal components
Figure 7. Click and drag on the Scree plot to zoom in and see the first set of principal components
- Mouse over the Scree plot to identify the point where additional PCs offer little additional information (Figure 8)
In this data set, a reasonable cut-off could be set anywhere between around 10 and 30 PCs. We will use 15 in downstream steps.
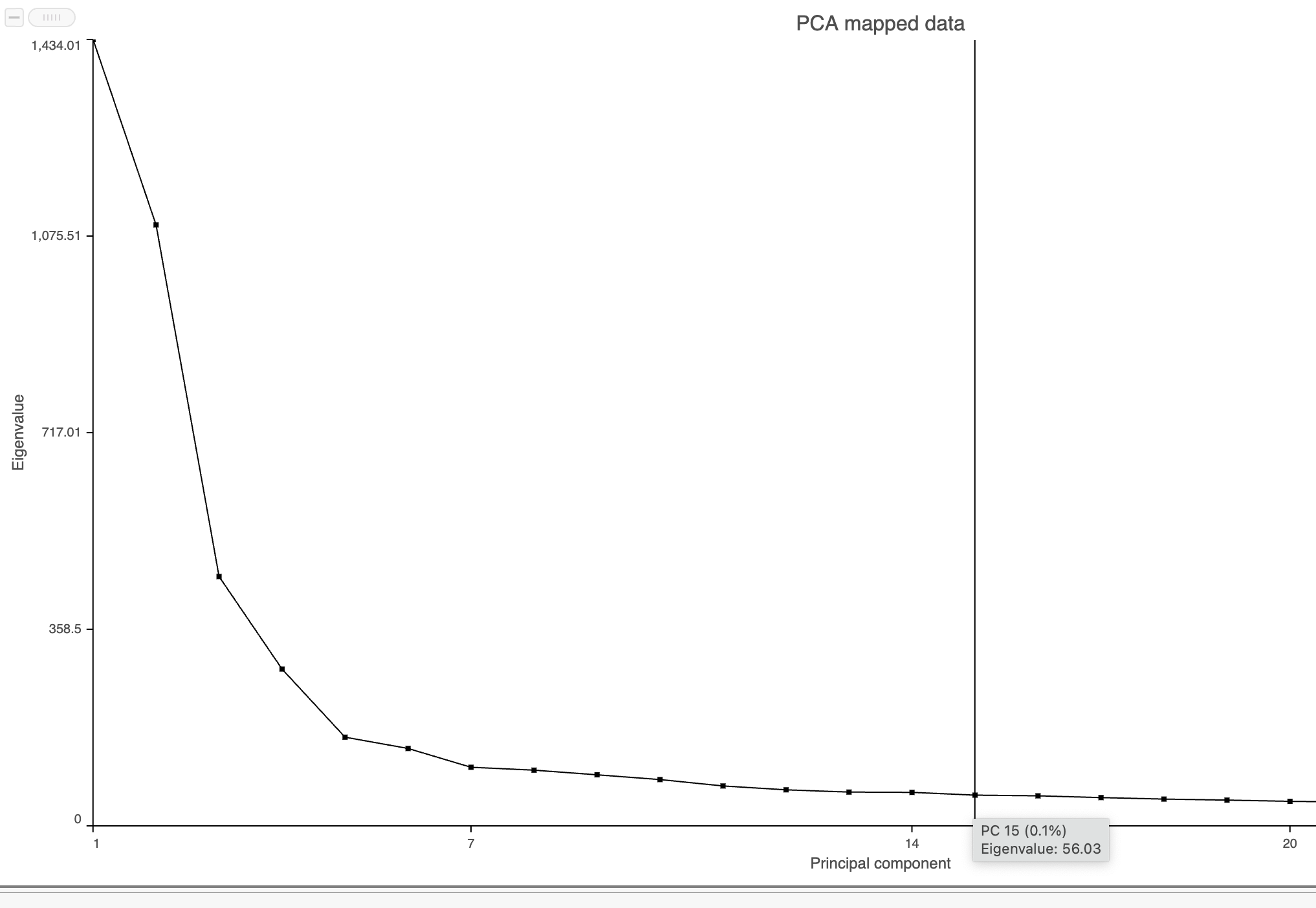 Figure 8. Identifying the optimal number of PCs
Figure 8. Identifying the optimal number of PCs
Graph-based clustering
We can use Graph-based clustering to group similar cells together in an unsupervised manner.
- Click the project name near the top to go back to the Analyses tab
- Click the circular PCA data node
- Click Exploratory analysis in the toolbox
- Click Graph-based clustering
- Click to Compute biomarkers
- Set the number of principal components to 15 (Figure 9)
- Click Configure under Advanced options and change the Resolution to 1.0
- Click Finish to run the task
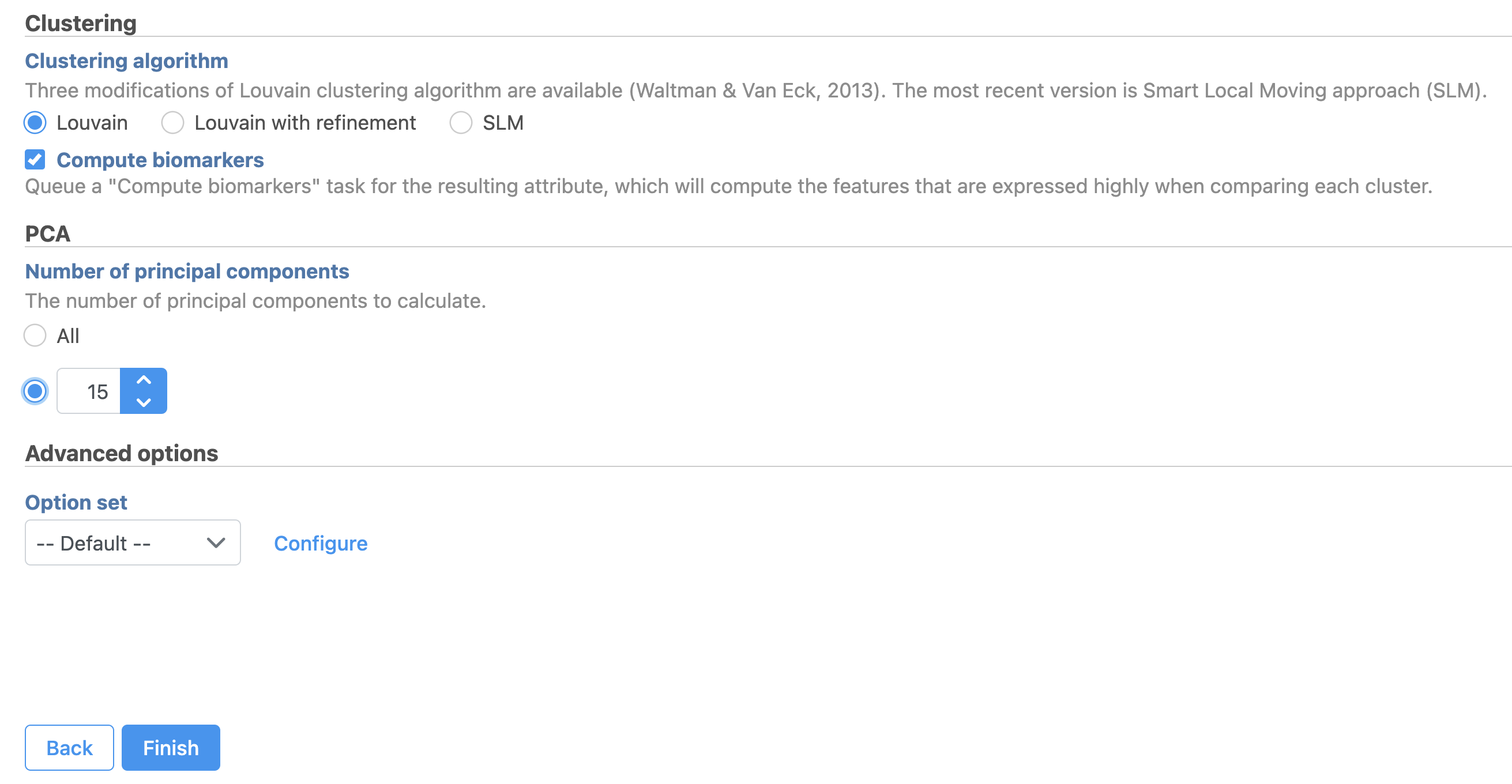 Figure 9. Graph-based clustering task set up. Reduce the number of PCs to 15
A Graph-based clustering task node will be added to the pipeline under the Analyses tab and a circular Graph-based clusters output data node will be produced (Figure 10)
Figure 9. Graph-based clustering task set up. Reduce the number of PCs to 15
A Graph-based clustering task node will be added to the pipeline under the Analyses tab and a circular Graph-based clusters output data node will be produced (Figure 10)
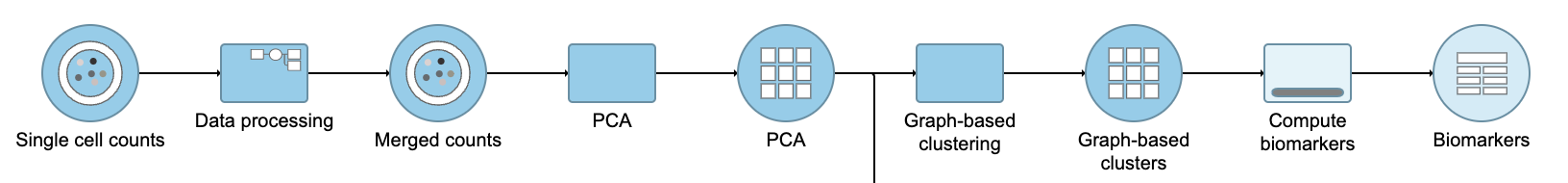 Figure 10. Graph-based clustering task and output data nodes
Figure 10. Graph-based clustering task and output data nodes
UMAP
Once the graph-based clustering task has completed, we can visualize the results with a UMAP plot. You could use the same steps here to generate a t-SNE plot. For this tutorial, we will use UMAP, as it is faster on several thousand cells.
- Click the circular PCA data node
- Click Exploratory analysis in the toolbox
- Click UMAP
- Set the number of principal components to 15 (Figure 11)
- Click Finish to run the task
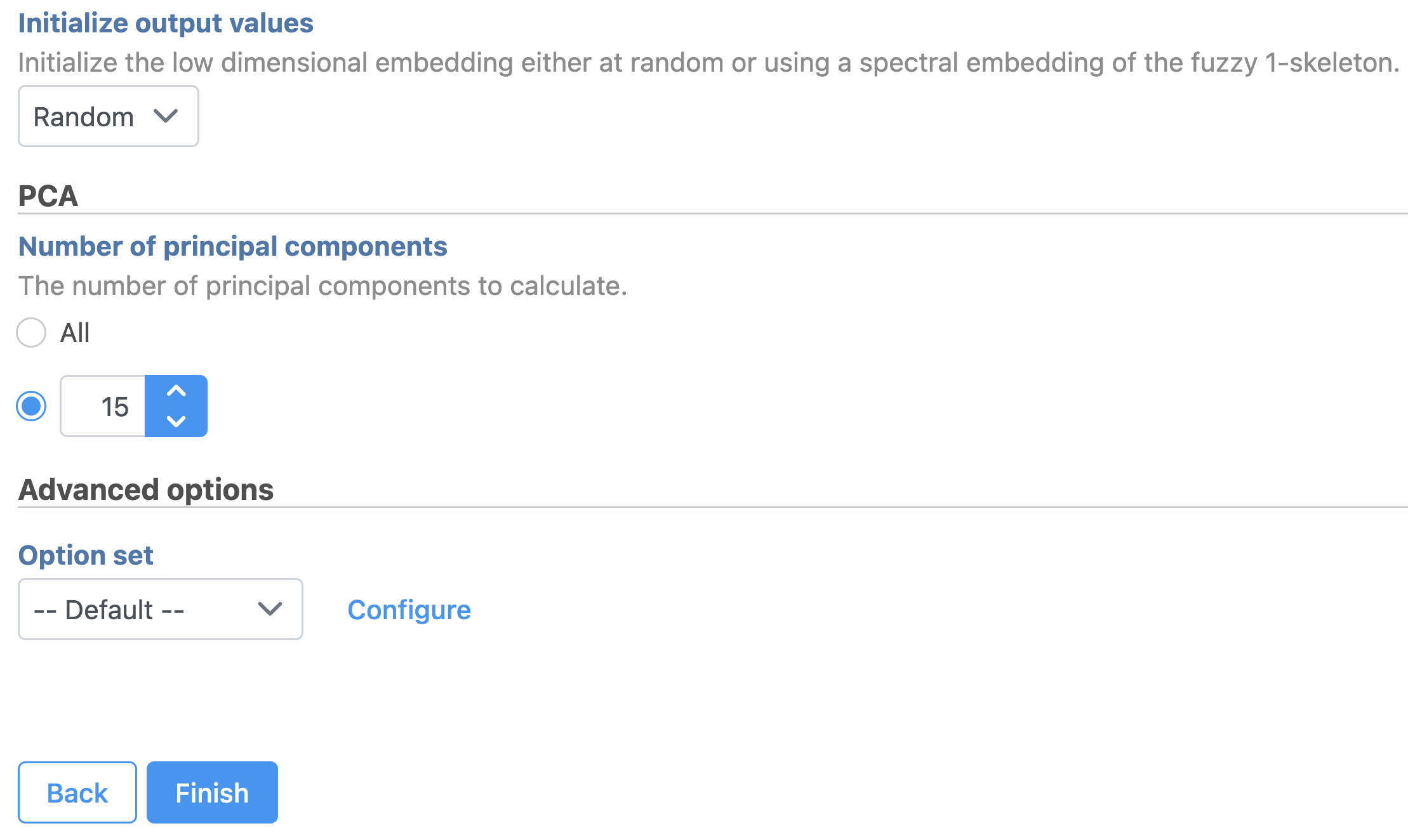 Figure 11. UMAP task set up. Reduce the number of PCs to 15.
A UMAP task node will be added to the pipeline under the Analyses tab and a circular UMAP output data node will be produced (Figure 12)
Figure 11. UMAP task set up. Reduce the number of PCs to 15.
A UMAP task node will be added to the pipeline under the Analyses tab and a circular UMAP output data node will be produced (Figure 12)
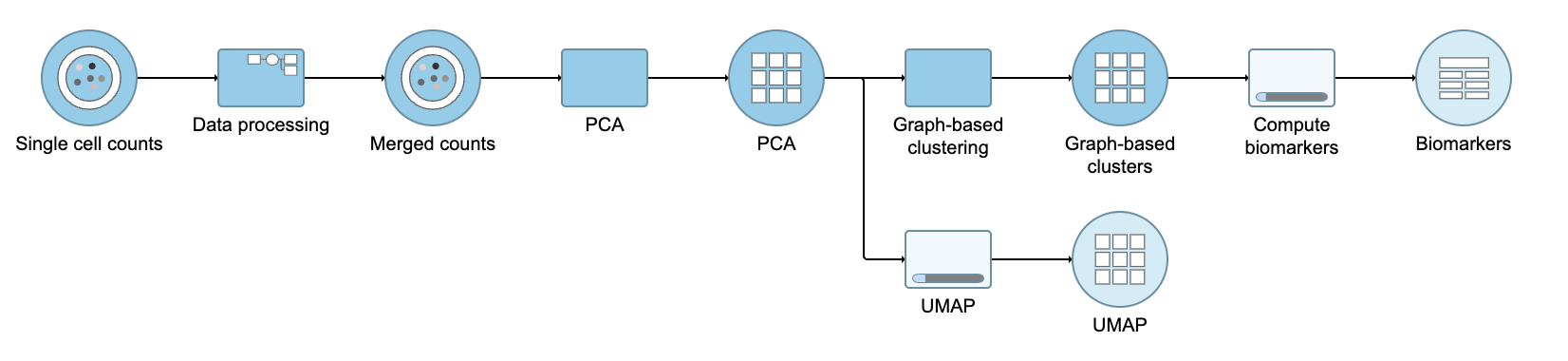 Figure 12. UMAP task and output data node
Figure 12. UMAP task and output data node
Notes on Performing Exploratory Analysis with Protein or Gene Expression Data Only
In this tutorial, we have performed exploratory analysis on merged protein and gene expression data, and we will perform classification on the merged data in the next step.
It can be interesting to perform exploratory analysis on the two feature types separately. For example, you might be interested to see how the clustering of the same cells differs between protein expression profiles vs. gene expression profiles.
To perform exploratory analysis on the two feature types separately, select the Merged counts data node, click Pre-analysis tools, followed by Split by feature type from the toolbox. A new task, Split by feature type, will be added to the pipeline resulting in two output data nodes: Antibody capture (protein data) and Gene expression (mRNA data). Both contain the same high-quality cells.
Performing exploratory analysis with gene expression data is the same as for the merged counts. Because there are a large number of genes, you will need to reduce the dimensionality with PCA, choose an optimal number of PCs and perform downstream clustering and visualization (e.g. graph-based clustering and UMAP/t-SNE). Performing exploratory analysis with protein data is different. There is no need to reduce the dimensionality as there are only a handful of features (17 proteins in this case), so you can proceed straight to downstream clustering and visualization. Figure 13 shows an example of how the pipeline might look if the data is split and analyzed separately.
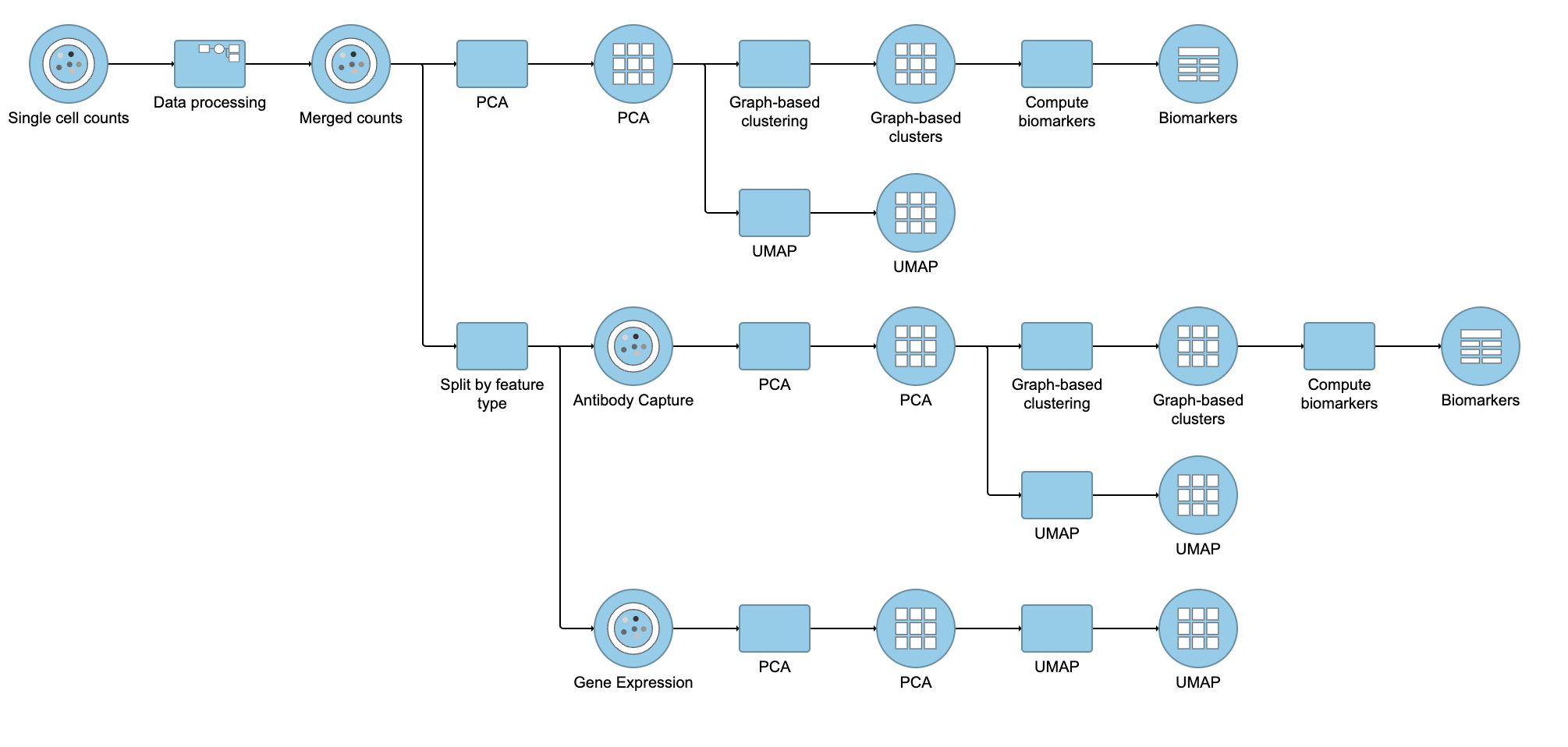 Figure 13. Example of how the pipeline might look if you split the merged counts and perform exploratory analysis for protein and gene expression data separately
You can then use the Data viewer to bring together multiple plots for comparison (Figure 14).
Figure 13. Example of how the pipeline might look if you split the merged counts and perform exploratory analysis for protein and gene expression data separately
You can then use the Data viewer to bring together multiple plots for comparison (Figure 14).
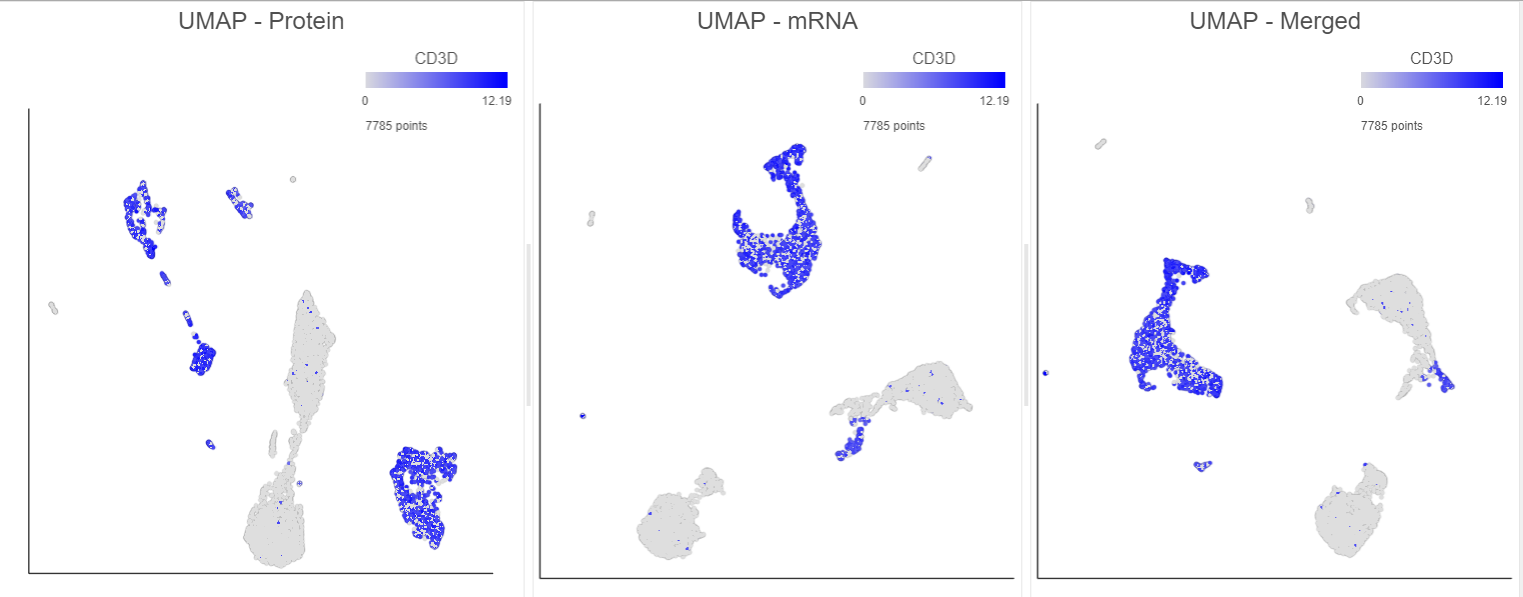 Figure 14. Comparison of 2D UMAP plots for the same cells clustered on protein, mRNA and merged data. All cells are coloured based on their expression of the CD3D gene (in blue). Note, the plots in this figure may differ from the default UMAP plots because these are 2D plots. Default UMAP plots re in 3D.
Figure 14. Comparison of 2D UMAP plots for the same cells clustered on protein, mRNA and merged data. All cells are coloured based on their expression of the CD3D gene (in blue). Note, the plots in this figure may differ from the default UMAP plots because these are 2D plots. Default UMAP plots re in 3D.
Additional Assistance
If you need additional assistance, please visit our support page to submit a help ticket or find phone numbers for regional support.


| Your Rating: |
|
Results: |

|
9 | rates |
Overview
Content Tools