We can check the quality of the samples using Partek Genomics Suite before analyzing the data.
Strand cross-correlation
In ChIP-Seq, genomic DNA is fragmented and target-protein-bound DNA fragments are purified by immunoprecipitation. These purified fragments are between 100 and 500 base pairs depending on the protocol; however, because ChIP-Seq uses short-read sequencing (25 to 35 base pair reads) to maximize sequencing depth, only the ends of each fragment will be sequenced. Consequently, with single-end sequencing, the forward and reverse strands for the each fragment will be from opposite ends of the fragment. At a protein-binding site, there will be two peaks of read enrichment, one from enrichment of forward strand reads and another from enrichment of reverse strand reads. The average distance between these peaks is termed the effective fragment length. Because the forward and reverse strand peaks are generated from a common set of fragments, the peaks should be roughly symmetrical. By phase shifting the data to the mid-point between the two peaks, a common read density plot can be created that shows single peaks at binding sites.
Strand Cross-Correlation allows us to use the symmetrical distribution of forward and reverse strand fragments calculate the effective fragment length (Kharchenko et al., 2008). The Pearson correlation coefficient between the read densities of the forward and reverse strands is calculated after phase shifts of between 0 and 500 base pairs. This is visualized with the phase shift range on the x-axis and the corresponding Pearson correlation coefficients between forward and reverse strand read densities on the y-axis (Figure 1). High-quality ChIP-Seq data will give a strong peak on the Strand Cross-Correlation plot at the effective fragment length. When calling peaks, the forward and reverse (or paired end) reads are each phase-shifted by the effective fragment length to create a combined read density profile.
For paired-end sequencing, Strand Cross-Correlation is calculated from the distribution of distances between the paired reads from the ends of each fragment.
We will perform Strand Cross-Correlation to identify the effective fragment length we can use when calling read enrichment peaks.
- Select Strand Cross-Correlation from the QA/QC section of the ChIP-Seq workflow
If you have not run this step before, you will be asked if you would like to create a new QA/QC child spreadsheet.
- If prompted, select Yes to create a new child spreadsheet for QA/QC
After running Strand Cross-Correlation, the Strand Separation of Samples viewer will open as a new tab (Figure 1).
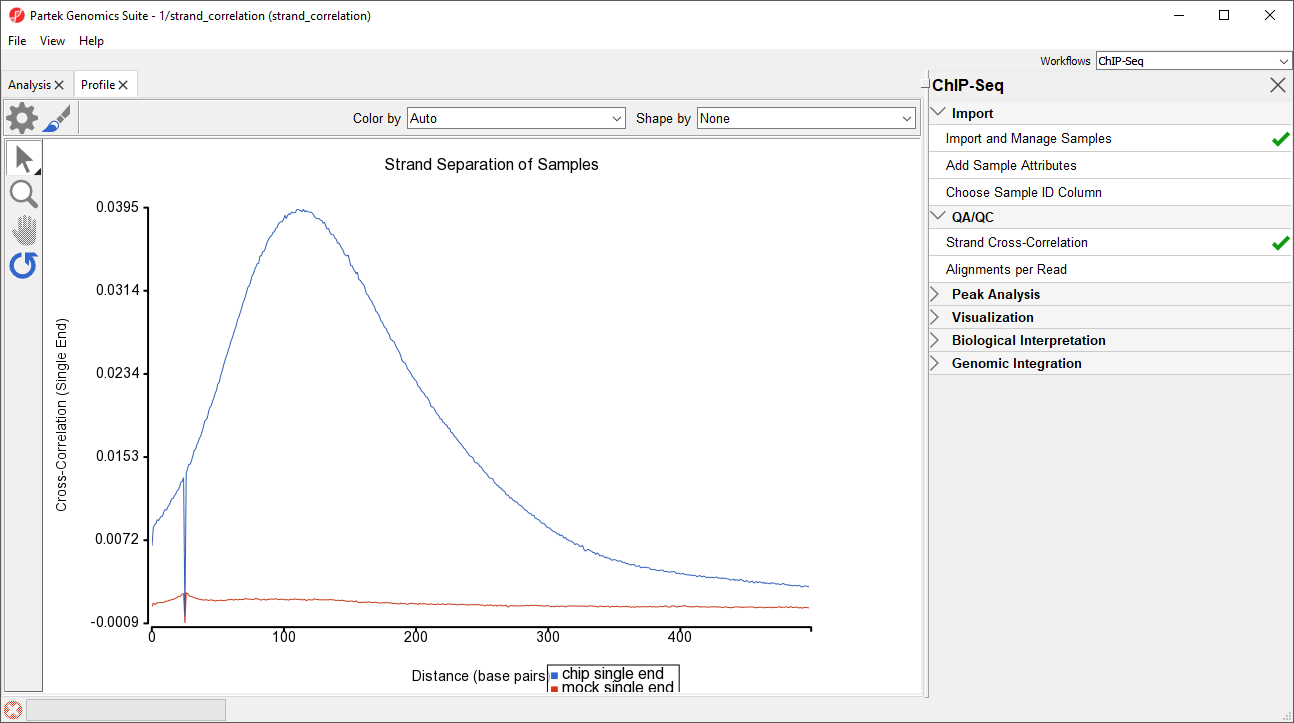
Figure 1. Strand Cross-Correlation profile plot showing possible effective fragment lengths on the x-axis and resulting Pearson correlation coefficients on the y-axis.
For the chip sample (blue), we can see the peak at 111 base pairs, corresponding to an effective fragment length of 111 base pairs. This number can be determined by examining the values in the strand_correlation spreadsheet (Figure 2), by moving the cursor over the peak in the graph, or by sorting the data in the spreadsheet. The Strand Separation of Samples graph is also useful as a quality control measure. In lower quality ChIP-Seq data, we would also observe a peak at the read length. The ratio between the Pearson correlation coefficient of the effective fragment length peak and the read length peak, normalized with the minimum correlation coefficient, [cc(fragment length) - min(cc)] / [cc(read length) - min(cc)] should be greater than 0.8 to meet the minimum quality standards recommended by the ENCODE project (Landt et al., 2012).
The mock sample (red) does not have an effective fragment length peak because it does not read density peaks to phase shift. It does have a small peak at the sequencing read length of 26 base pairs.
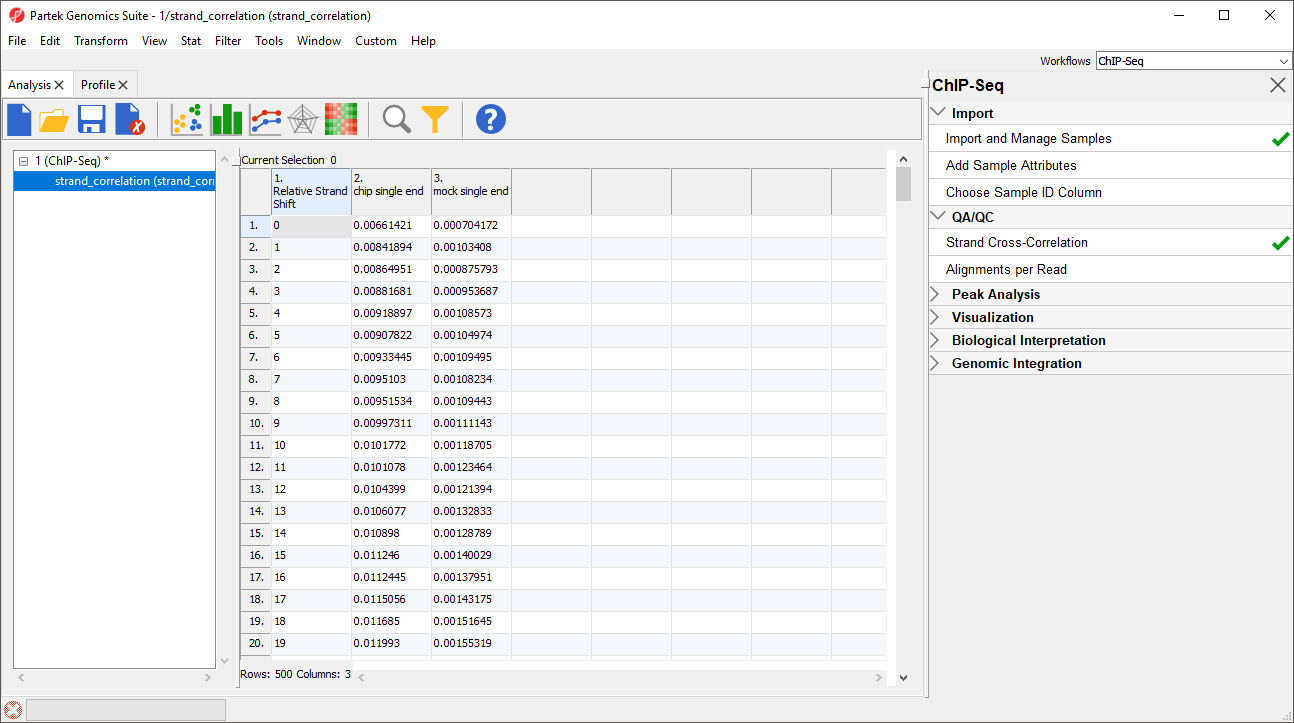
Figure 2. The strand correlation spreadsheet shows the Pearson correlation coefficients for each relative strand shift value (effective fragment length)
Checking the distribution of reads
BAM files can contain both aligned and unaligned reads. The spreadsheet created during import shows the number of reads that were aligned to the reference genome. A large number of unaligned reads may be the result of poor quality sequencing data or alignment problems. It may also be useful to know how many reads map to more than one location in the genome if the options used during alignment supported multiple-mapped reads.
- Select Alignments per read form the QA/QC section of the ChIP-Seq workflow
A new spreadsheet named Alignment_Counts will be generated (Figure 3).
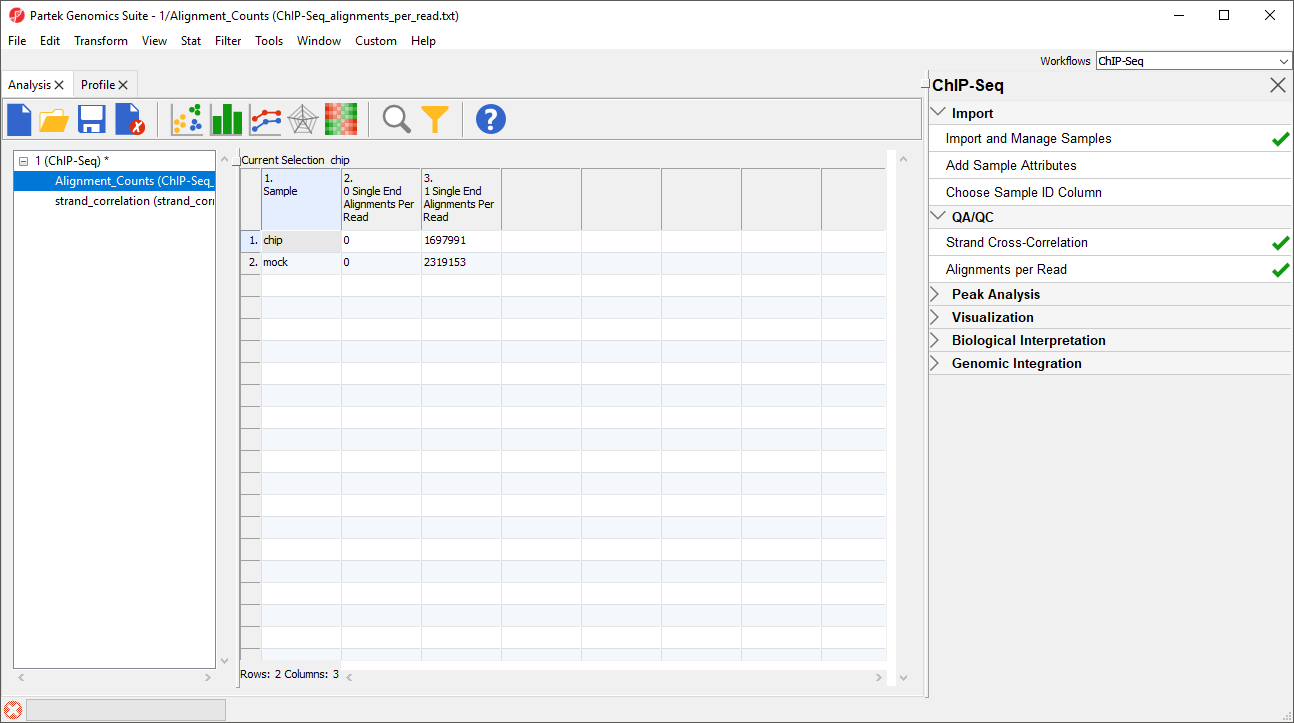
Figure 3. Unaligned reads have been removed from these BAM files and the alignment options did not permit mapping to more than one location
The titles of columns 2. 0 Single End Alignments Per Read and 3. 1 Single End Alignment Per Read indicate that this is single end data. Column 2 shows the number of unaligned reads, while column 3 shows the number of reads that aligned exactly once. If the BAM files used in this tutorial included reads that mapped to more than one location in the genome, there would be additional columns.
Additional Assistance
If you need additional assistance, please visit our support page to submit a help ticket or find phone numbers for regional support.


| Your Rating: |
|
Results: |

|
35 | rates |
Overview
Content Tools