Data for this tutorial can be downloaded from the Partek website using this link - ChIP-Seq tutorial data. To follow this tutorial, download the two .bam files and unzip them on your local computer using 7-zip, WinRAR, or a similar program.
- Store the two.bam files at C:\Partek Training Data\ChIP-Seq or to a directory of your choosing. We recommend creating a dedicated folder for the tutorial on a local drive.
- Select ChIP-Seq from the Workflows drop-down menu (Figure 1)
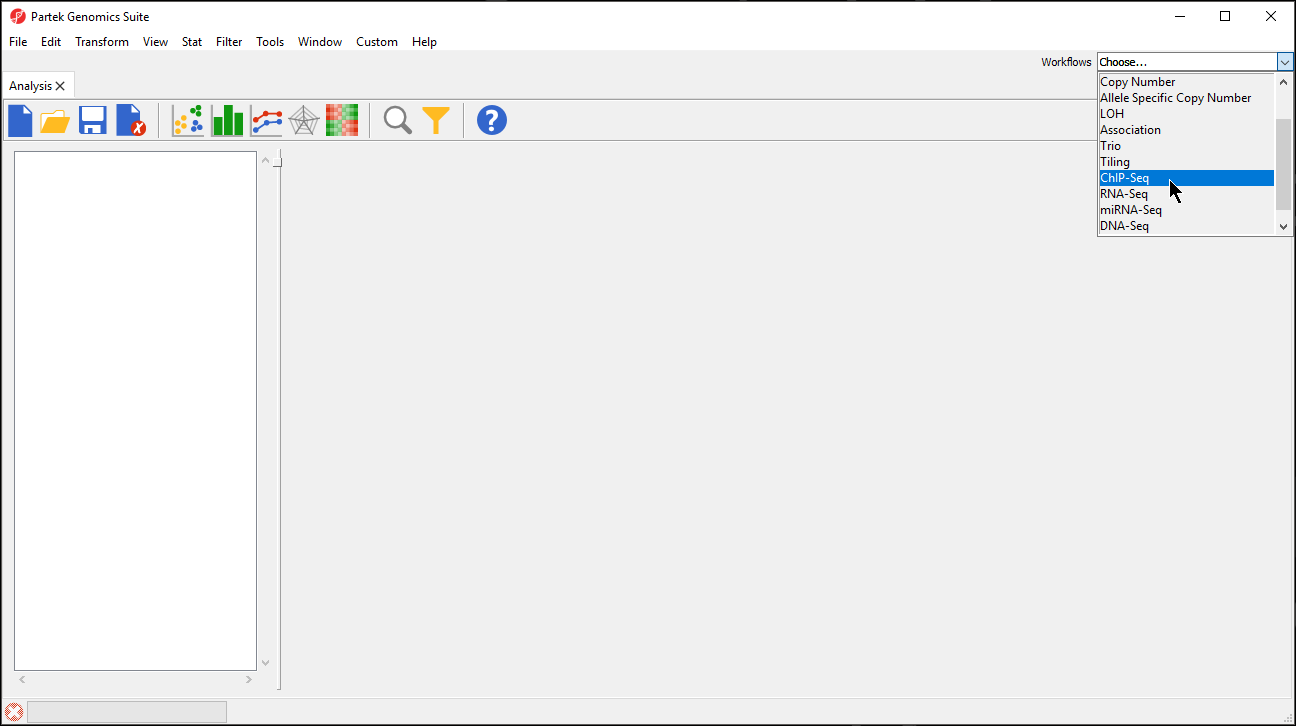
Figure 1. Selecting the ChIP-Seq workflow
- Select Import and Manage Samples from the Import section of the ChIP-Seq workflow
- Select Browse... or use the file tree to navigate to the folder where you stored the .bam files
All .bam files in the folder will be selected by default (Figure 2).
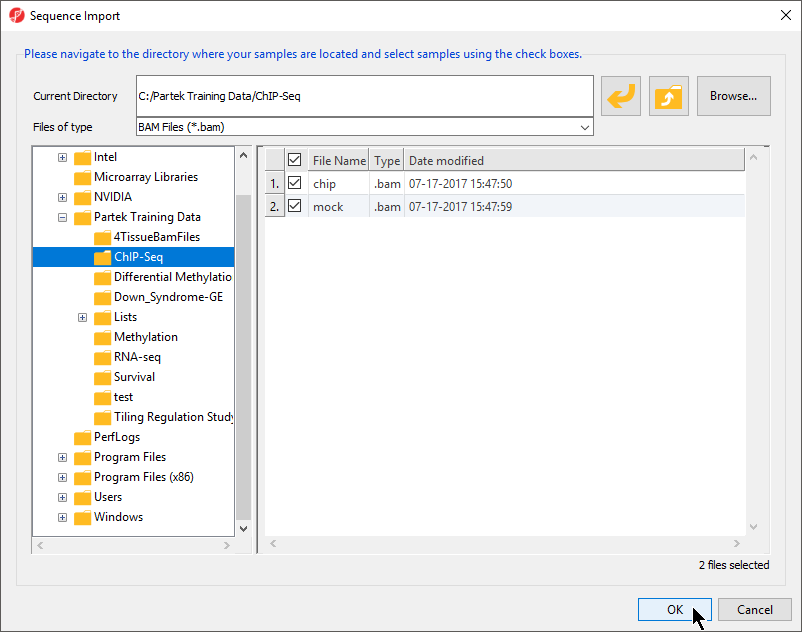
Figure 2. Importing .bam files using the Sequence Import dialog
- Verify that chip.bam and mock.bam are selected
- Select OK
The Sequence Import dialog will launch (Figure 3). This allows us to choose the output file name and destination for the parent spreadsheet, as well as the species, and genome build of the imported samples. By default, the output file destination is the folder the .bam files are located.
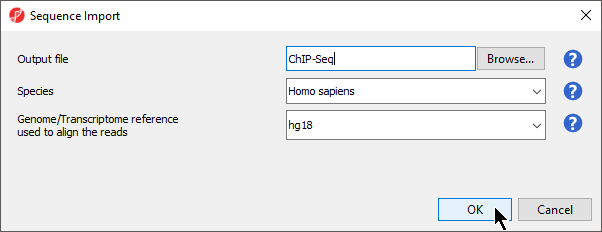
Figure 3. Setting the output file name, species, and genome build during .bam file import
- Set Output file to ChIP-Seq
- Set Species to Homo sapiens using the drop-down menu
- Set Genome build to hg18 using the drop-down menu
- Select OK
The Bam Samples Manager dialog will open (Figure 4). This dialog can be used to add samples to the project (Add samples), remove samples (Remove samples), to associate multiple files with particular samples (Manage samples), and to map the chromosome names from the input files to the association files (Manage sequence names).
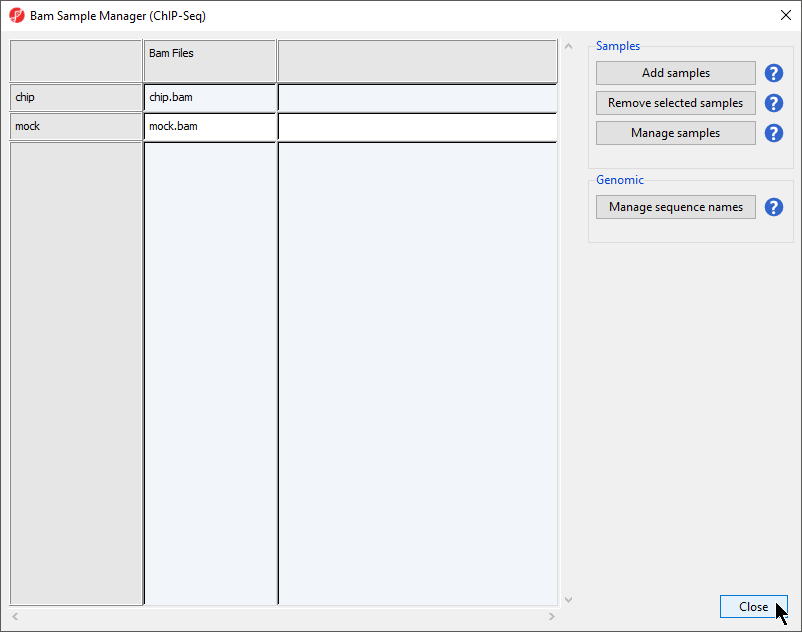
Figure 4. The Bam Sample Manager can be used to add, remove, and manage files and samples
- Select Close
The Sort bam files dialog will open. Sorting is necessary for all imported .bam files, but you can choose to hide this hint in the future by selecting Please don't show me this hint again.
- Select OK
The imported spreadsheet will open while the .bam files are sorted. Progress in sorting will be displayed on the progress bar in the lower left-hand corner of the Partek Genomics Suite window. Once sorting has completed, there will be samples on rows with the sample names in column 1. Sample ID and the number of reads mapped to the reference genome for each sample in column 2. Number of allignments (Figure 5).
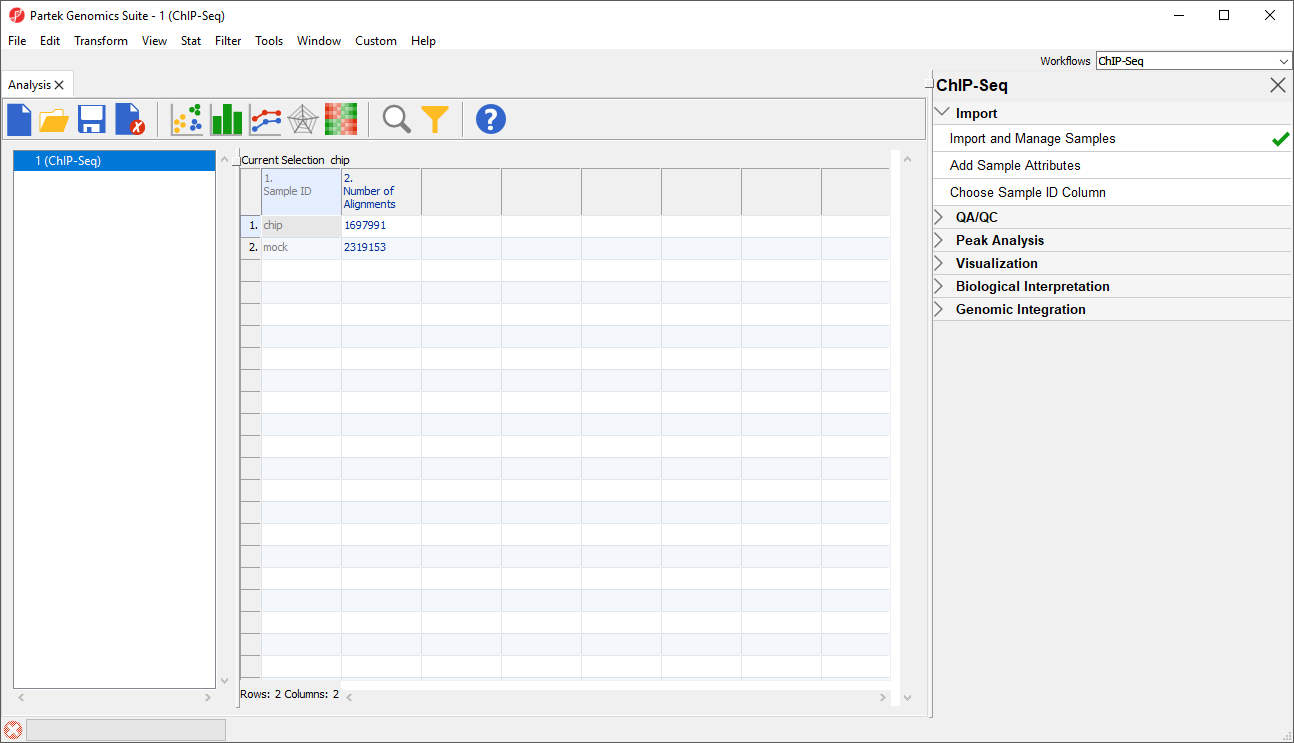
Figure 5. Imported .bam files with one sample in each row
Additional Assistance
If you need additional assistance, please visit our support page to submit a help ticket or find phone numbers for regional support.


| Your Rating: |
|
Results: |

|
36 | rates |
Overview
Content Tools