SAMtools1 (version 1.2) utilizes the mpileup command to look at observed bases in the reads covering every genomic position represented in the aligned sequence data and calculate the likelihood of every possible genotype at a locus. Subsequently, bcftools applies the prior probability and uses Bayesian inference to call actual genotypes, outputting variant information in Variant Call Format (vcf). This method can identify both single nucleotide variants and insertions/deletion events. General information about the underlying algorithm utilized by SAMtools is detailed by Li. 2,3
SAMtools dialog
Selecting SAMtools from the context sensitive menu will bring up the SAMtools task dialog, which contains three default sections: Variant detection method, Select Reference sequence, and Advanced options.
In the Variant detection method drop-down list, Against reference will compare base composition for each sample against the reference sequence assembly, independently (Figure 1).
 Figure 1. Selecting a variant detection method in the Samtools dialog
Figure 1. Selecting a variant detection method in the Samtools dialog
Selection of Among samples (in the absence of sample attributes or when Paired analysis is not selected) utilizes information from all samples in the project in a joint variant detection. Against reference typically performs well when samples have good coverage and may contain unique SNPs. In instances where there is low coverage, Among samples may perform well for identifying SNPs shared between samples (but not for unique SNPs).
In the event paired samples exist within the project, detection Among samples can be utilized to identify loci with differing genotypes between the pair once each sample has been compared to the reference sequence assembly. In instances where there is limited information to accurately determine genotypes in one or both of the samples, the same genotype may be called for case and control if it differs from the reference. The Filter variants task can be used to exclude these spurious loci. To perform this analysis, sample attributes must be added in the Data tab of the project (Figure 2). Specifically, an attribute must be added for sample ID (shared between the paired samples) and an attribute must also be added for sample type that differentiates the paired samples. Examples of the latter can include case and control or tumor and normal. If these attributes are present, a section for Analysis options will be displayed below the Variant detection method (Figure 3). To utilize this feature, select Paired analysis. Match ID must then be specified and should correspond to the attribute that references the sample ID shared between the pair. Selecting Case/control will allow for discriminating genotypes between paired samples in downstream tasks. Attribute should correspond to the attribute that defines type within sample pairs, and Control can be specified for whatever category relates to the reference sample.
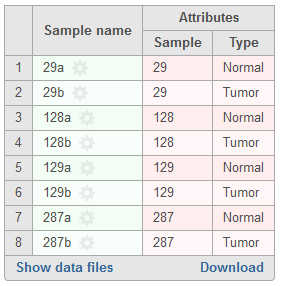 Figure 2. Example of attributes required for paired variant analysis
Figure 2. Example of attributes required for paired variant analysis
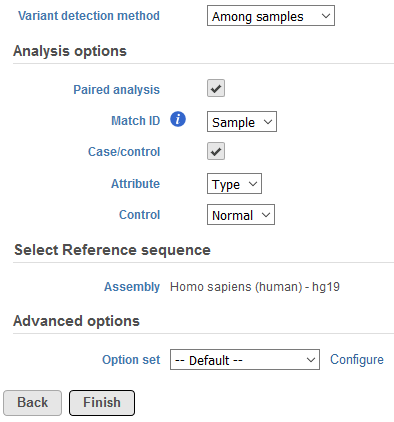 Figure 3. Specifying options for paired variant detection
Figure 3. Specifying options for paired variant detection
Select Reference sequence will specify the reference assembly to utilize for variant detection. If the alignment was generated in Partek® Flow®, the Assembly will be displayed as text in the section, and you do not have the option to change the reference. In the event that alignment was performed outside of Partek Flow, you will need to select the appropriate Assembly utilized for alignment in the drop-down list. Assemblies previously added to library files (see Library File Management) will be available for selection or New assembly… can be utilized to import the reference sequence to library files from within the task.
Advanced options provides a means to tune parameters in the variant detection for optimal performance. Upon invoking the task dialog, Option set is set to Default, and these parameters are provided by the SAMtools developers. Clicking Configure will open a window to tune advanced options (Figure 3). Moving the mouse cursor over the info button![]() will provide details for each parameter. Please refer to the SAMtools documentation for further details on any of these parameters.
will provide details for each parameter. Please refer to the SAMtools documentation for further details on any of these parameters.
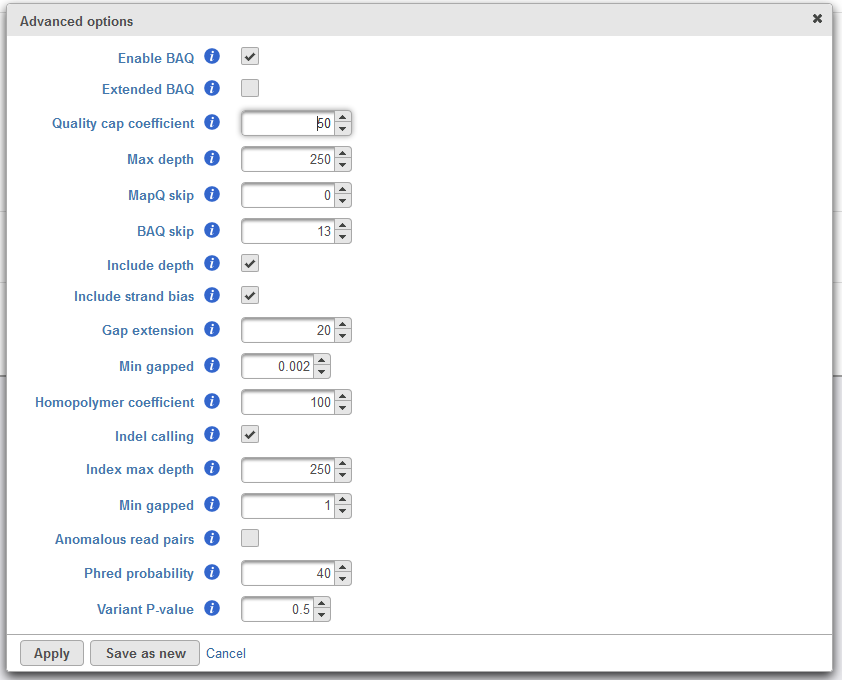 Figure 4. Configuring advanced Samtools options
Figure 4. Configuring advanced Samtools options
References
- Li H, Handsaker B, Wysoker A, et al. The Sequence Alignment/Map format and SAMtools. Bioinforma Oxf Engl. 2009;25(16):2078-2079.
- Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27(21):2987-2993.
- Li H. Improving SNP discovery by base alignment quality. Bioinformatics. 2011;27(8):1157-1158.
Additional Assistance
If you need additional assistance, please visit our support page to submit a help ticket or find phone numbers for regional support.


| Your Rating: |
|
Results: |

|
39 | rates |
Overview
Content Tools
1 Comment
Melissa del Rosario
author: eseiser