Variant detection can identify large numbers of variants, dependent both on the size of the regions being interrogated and the parameters utilized during detection. As such, filtering variants is often a necessary task to aid in identifying variants that may be relevant for downstream investigation. Partek Flow provides tools for the filtering variant data both in regards to quality metrics generated during detection and annotation information. The task can be invoked from any Variants or Annotated variants data node.
Filter variants dialog
The Filter variants task dialog can contain two to five sections, dependent on the variant caller used for detection and the level of annotation (Figure 1). All instances of the task will include the following: Include region overlapping variants and a section for Quality.
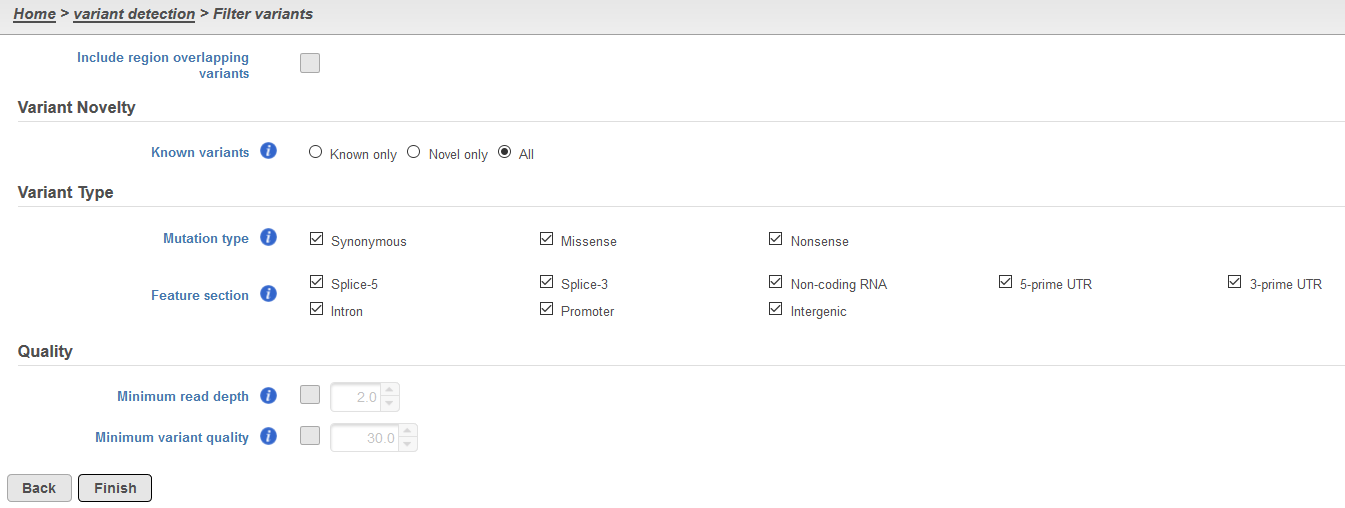 Figure 1. Components of the Filter variants dialog
Figure 1. Components of the Filter variants dialog
Selecting Include region overlapping variants will bring up a dialog to include variants located within genomic regions of interest (Figure 2), which could be regions such as transcript models or amplicons. If variant detection was performed in Partek® Flow®, the Assembly will be displayed as text in the section, and you do not have the option to change the reference. In the event that variant detection was performed outside of Partek® Flow®, you will need to select the appropriate Assembly utilized for variant detection in the drop-down list. Assemblies previously added to library files (see Library File Management) will be available for selection or New assembly… can be utilized to import the reference sequence to library files from within the task. The Gene/feature section will allow for the use of any annotation model specified in the library files (see Library File Management) in the drop-down menu or can be imported from within the task by selecting Add annotation model. If an annotation that contains gene-level information is selected, this filter will include both intronic and exonic regions.
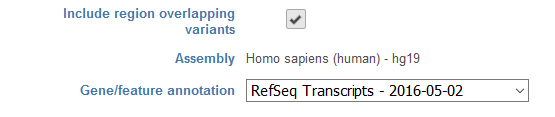 Figure 2. Filtering with Include region overlapping variants
Figure 2. Filtering with Include region overlapping variants
If the filter is invoked from an Annotated variants data node, the Variant Novelty section can be utilized to filter known variants as identified in a variant database used for annotation (Figure 3). Selecting Known only, Novel only, or All will include only these types of variants in the resulting filtered variants.
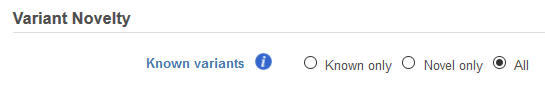 Figure 3. Filtering with Variant Novelty for data annotated with a variant database
Figure 3. Filtering with Variant Novelty for data annotated with a variant database
Variants annotated with a transcript model will include a filter for Variant Type (Figure 4). For variants in coding regions, Mutation type allows for the inclusion of Synonymous, Missense, and/or Nonsense variants when selecting the appropriate type. For variants located outside of coding regions, Feature section allows for the inclusion of 5-prime splice site (Splice-5), 3-prime splice site (Splice-3), Non-coding RNA, 5-prime UTR, 3-prime UTR, Intron, Promoter, and/or Intergenic variants by selecting the appropriate type.
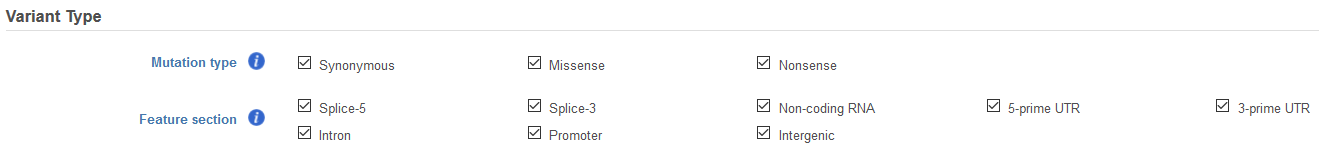 Figure 4. Filter with Variant Type for data annotated with a transcript model
Figure 4. Filter with Variant Type for data annotated with a transcript model
The Allele Frequency section is available for variants detected using LoFreq and provides a means to identify low frequency variants within samples by excluding variant alleles that are present above the frequency specified for Maximum estimated allele frequency (Figure 5). This may be useful for determining variants identified in samples with heterogeneous DNA populations, such as cell-free DNA.
 Figure 5. Filtering by Allele frequency derived from LoFreq variant detection
Figure 5. Filtering by Allele frequency derived from LoFreq variant detection
The Quality section of the filter task will involves metrics generated from the variant detection, and these will be dependent upon the method utilized for variant detection. Moving the mouse cursor over the info button![]() will provide details for each parameter. Decisions on quality filtering parameters should be based upon sequencing assay design as well as goal or the study, either identification of all potential variants or identification of high confidence variants. At the very least, the use of Minimum read depth should be considered for filtering to ensure sufficient read evidence was available to call a variant. In instances where paired variant detection was performed in SAMtools, Minimum genotype log ratio may be employed to ensure sufficient evidence of genotype differences in case and control sample pairs. Please refer to the Samtools, FreeBayes, and LoFreq documentation for further details on any of these parameters.
will provide details for each parameter. Decisions on quality filtering parameters should be based upon sequencing assay design as well as goal or the study, either identification of all potential variants or identification of high confidence variants. At the very least, the use of Minimum read depth should be considered for filtering to ensure sufficient read evidence was available to call a variant. In instances where paired variant detection was performed in SAMtools, Minimum genotype log ratio may be employed to ensure sufficient evidence of genotype differences in case and control sample pairs. Please refer to the Samtools, FreeBayes, and LoFreq documentation for further details on any of these parameters.
 Figure 6. Example of filtering variants based on Samtools quality metrics
Figure 6. Example of filtering variants based on Samtools quality metrics
Additional Assistance
If you need additional assistance, please visit our support page to submit a help ticket or find phone numbers for regional support.


| Your Rating: |
|
Results: |

|
40 | rates |
Overview
Content Tools
1 Comment
Melissa del Rosario
author: eseiser