The library files associated with the selected assembly are organized into several sections. Below is some information on each section.
Reference Files
This section includes two types of library file: reference sequence and cytoband files.
Reference sequences are the chromosome/scaffold/contig DNA sequences for a species. A reference sequence file is typically in FASTA or 2bit format. The reference sequence of a species is used for aligner index creation, variant detection and visualization of the reference sequence in the Chromosome view.
Cytoband files are used for drawing ideograms of chromosomes in the Chromosome view, including positions of cytogenetic bands if known.
Reference aligner indexes
Next-generation sequencing aligners require the reference sequence to be indexed prior to alignment, as this greatly increases alignment speed. An index consists of a set of files (Figure 1) and are generally aligner specific. For example, if you wish to align using BWA, you need a BWA index.
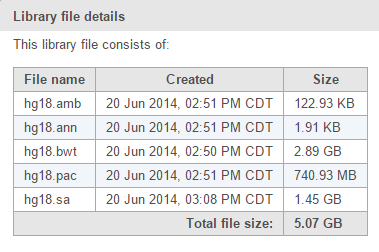
Figure 1. BWA reference aligner index files for human hg18 assembly
Some of the supported aligners share indexes. If you want to align using Tophat, the Bowtie aligner indexes can be used. If you want to align using Tophat2, the Bowtie2 aligner indexes can be used.
Some aligner indexes are version specific, so care must be taken if you change aligner versions. For example, the index files for STAR version 2.4.1d are different to older versions of STAR.
This section contains aligner indexes for aligning to the whole genome. If you wish to align to a subset of the genome, e.g. targeted amplicons or the transcriptome, you must generate these indexes in the Annotation models section.
Gene sets
Gene set files are required for biological interpretation analyses (e.g. GO enrichment). Genes are grouped together according to their biological function. Gene set files have to be in GMT format, where each row represents one gene set. The first column of a GMT file is the GO ID or gene set name. The second column is an optional text description. Subsequent columns are the gene symbols that belong to each gene set. Gene ontologies for various model organisms are available for automatic download from the Partek repository (source: geneontology.org). Because gene ontologies are frequently updated, geneontology.org is checked for updates quarterly. You can check for recent updates to the Partek repository here.
Variant annotations
Variant annotation databases are collections of known genomic variants (e.g. single nucleotide polymorphisms). If you have performed a variant detection study, detected variants can be searched against variant annotation library files to see if the detected variants are known from previous studies. Furthermore, you can validate detected variants against 'gold-standard' variant annotation library files. Variant annotation files are typically in VCF format.
Variant annotation databases from commonly used sources (e.g. dbSNP) are available for automatic download from the Partek repository. Because variant annotation databases are frequently updated, these sources are checked for updates quarterly. You can check for recent updates to the Partek repository here.
SnpEff variant databases
SnpEff1 is a variant annotation and effect prediction tool that requires its own variant annotation files, separate to the other Variant annotation library files. If you wish to use SnpEff, library files need to be added to this section.
VEP database
The Ensembl Variant Effect Predictor (VEP) is another variant annotation and prediction tool that requires its own annotation files, separate to the Variant annotation library files. If you wish to use VEP, library files need to be added to this section.
Annotation models
This section includes two types of library file: annotation models & aligner indexes.
Annotation models describe genomic features (e.g. genes, transcripts, microRNAs) for a specific version of the reference sequence. Annotation models contain labels (e.g. gene ID) and genomic coordinates (e.g. chromosome, start & stop position) for each feature.
Annotation models will appear in separate tables (Figure 2). If you have multiple versions of annotation models from the same source, it is advisable to distinguish them by their date or version number.
Annotation models from commonly used sources (e.g. Refseq, ENSEMBL) are available for automatic download from the Partek repository. Because annotation models are frequently updated, these sources are checked for updates quarterly. You can check for recent updates to the Partek repository here.
Annotation models are used for quantification in gene expression analyses, annotating detected variants (e.g. to predict amino acid changes), visualizations in Chromosome view, generating coverage reports and for aligner index creation (see Adding Aligner Indexes Based on an Annotation Model). Typical file formats include GTF, GFF, GFF3 and BED.
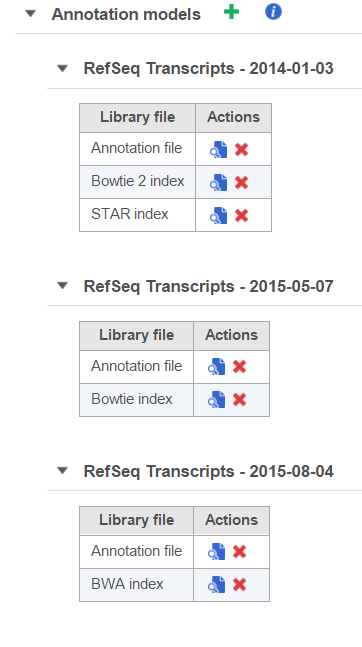
Figure 2. Annotation models are displayed in separate tables
The gray arrows (![]() /
/![]() ) next to the annotation model name expand/collapse each table. The three annotation models displayed in Figure 2 are different versions from the same source (RefSeq), distinguishable by their date. Aligner indexes (e.g. for alignment to the transcriptome) are added to the table of the corresponding annotation model.
) next to the annotation model name expand/collapse each table. The three annotation models displayed in Figure 2 are different versions from the same source (RefSeq), distinguishable by their date. Aligner indexes (e.g. for alignment to the transcriptome) are added to the table of the corresponding annotation model.
The aligner indexes in the Annotation models section are required if you wish to align to a subset of the genome as defined by the annotation model, e.g. target amplicons or the transcriptome. The reference sequence is still required to generate an aligner index for an annotation model. As with whole genome alignment, indexes are aligner specific, although some aligners share indexes and are version specific (see Reference aligner indexes). The aligner indexes generated will be added to the corresponding annotation model table (Figure 2).
References
- Cingolani P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 6(2):80-92. PMID: 2272867
Additional Assistance
If you need additional assistance, please visit our support page to submit a help ticket or find phone numbers for regional support.


| Your Rating: |
|
Results: |

|
39 | rates |
Overview
Content Tools