HTSeq is set of tools for processing high-throughput sequencing data1. In Partek Flow, we have implemented the htseq-count script from HTSeq for quantifying aligned reads to an annotation model.
The input for HTSeq is an Aligned reads data node and a Gene/Feature annotation file.
To run HTSeq:
- Click an Aligned reads data node
- Click the Quantification section of the toolbox
- Click HTSeq
Please note that HTSeq has not been optimized for performance and can take a very long time to run compared with Quantify to annotation model (Partek E/M) on the same data.
Configurable options
HTSeq includes basic options (Figure 1) and advanced options accessible by clicking Configure (Figure 2).
Basic Options
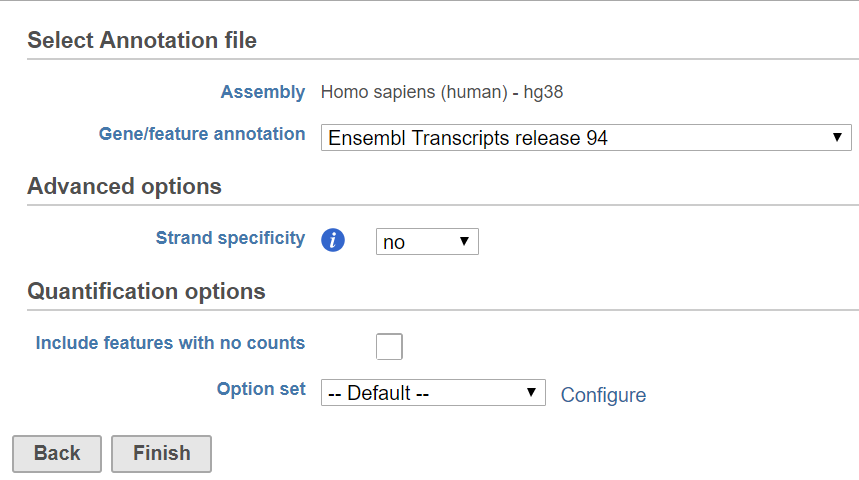
Figure 1. Basic HTSeq options
Annotation file
The annotation file contains the features the aligned reads will be quantified to. For more information about adding an annotation model, please see Adding an Annotation Model.
Strand specificity
Depending on the library preparation method, information about the strand of the original transcript may be faithfully preserved or lost. This setting controls whether HTSeq considers strand during quantification. Consult your library preparation method user manual if you are unsure about if and how the method preserved strand information.
If set to no, a read is considered to be overlapping a feature regardless of whether it maps to the same or opposite strand as the feature.
If set to yes, the read has to be matched to the same strand as the feature if single-end reads and matched to the same strand for the first read and the opposite strand for the second read if paired-end reads.
If set to reverse, the read has to be matched to the opposite strand as the feature if single-end reads and matched to the opposite strand for the first read and the same strand for the second read if paired-end reads.
Include features with no counts
By default, only features (e.g., genes) with one or more aligned read will be included in the output. If this option is selected, all features from the annotation model, including those without any matching aligned reads, will be included.
Advanced Options
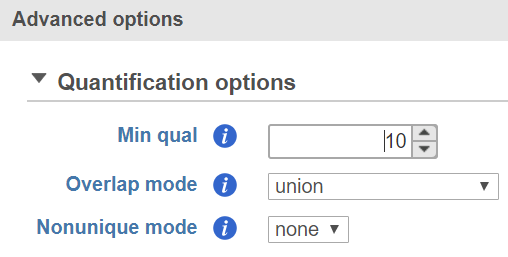
Figure 2. Advanced options for HTSeq
Min qual
HTSeq will skip reads with an alignment quality lower than the value specified here. The default is 10.
Overlap mode
This option determines how HTSeq handles reads that partially overlap features. The default is union.
If set to union, any read that is partially overlapped by a feature will be assigned to that feature. Assignment is non-exclusive if multiple feature overlap.
If set to intersection-strict, only reads that are fully overlapped by a feature will be assigned to that feature. Assignment is non-exclusive if multiple feature fully overlap.
If set to intersection-nonempty, reads are assigned to the feature that has the greatest overlap. Assignment is non-exclusive if multiple feature overlap the same amount.
Nonunique mode
This option determines how HTSeq counts reads that are assigned to more than one feature. The default is none.
If set to none, reads that are assigned to more than one feature are not counted for any feature.
If set to all, reads are counted for all features they are assigned to.
Output
HTSeq outputs a Gene counts data node (Figure 3). There is no task report.
Figure 3. HTSeq output
References
Simon Anders, Paul Theodor Pyl, Wolfgang Huber. HTSeq — A Python framework to work with high-throughput sequencing data. Bioinformatics (2014), in print, online at doi:10.1093/bioinformatics/btu638
Additional Assistance
If you need additional assistance, please visit our support page to submit a help ticket or find phone numbers for regional support.


| Your Rating: |
|
Results: |

|
25 | rates |
Overview
Content Tools