Data tracks section of the Select tracks dialog enables you to specify the tracks for visualization on the canvas. An overview of the available track types is provided in Figure 1. Note that not all tracks are visible at all times and that their presence depends on the zoom level. The tracks can be customised and their appearance changed by using the control panel on the right.
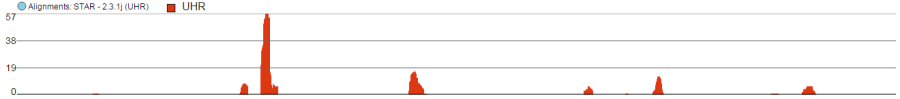
Alignments track
Isoform proportion track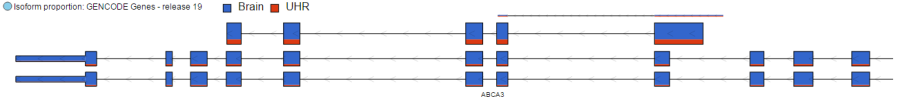
Variants track
Amino acids track
Reads pileup track
Probe intensities track
Figure 1. Data tracks in Chromosome view (examples)
Alignments Track
Alignments track displays a view of alignments present in .bam files in a stacked histogram fashion (similar to Partek® Genomics Suite®). The y-axis shows number of (raw) base calls per position. By default, reads are coloured by sample; the exception is invocation of the chromosome view on a variant table, when the reads are coloured by base calls. The difference is shown in Figure 2.
Reads coloured by sample
Reads coloured by base calls
Figure 2. Alignments track: different colouring options. When colouring reads by sample, the reads are stacked (on top of each other), i.e. in the example above there are more reads in the red sample than in the blue sample
Isoform Proportion Track
The Isoform proportion track displays the reads mapped to transcripts and helps to visualize differential expression and alternative splicing, using standard symbols for exons (boxes) and introns (lines connecting the boxes). The size of each transcript is proportional to the number of reads that map to that transcript. The color indicates the samples to which the reads belong. Figure 3 shows a gene with two transcripts in RefSeq database; the top transcript is more abundant than the bottom transcript and is preferentially expressed in the "blue" condition (labeled as 0 uM). The bottom transcript, on the other hand, seems to be expressed at the same level across all three conditions (i.e. 0 uM, 5 uM, 10 uM). The number and structure of transcripts on the plot depend on the transcript model that was used for mapping.
Figure 3. Isoform proportion track: the transcripts are shown as present in the transcript model that was used for mapping. Exons are depicted as boxes. The size of each transcript is proportional to the number of reads mapping to it. Colors indicate samples to which the reads belong
Variants Track
Variant tracks show single nucleotide variants (SNVs) and indels, and appear in the Select track dialog if Detect variants task has been performed. Presentation of variants depends on the level of zoom. With low power magnification, SNVs are seen as purple columns and indels are bars (insertions: green bars; deletions: red bars) (Figure 4).
Figure 4. Variants track at low power magnification: SNVs are symbolized by purple columns and an insertion is presented as a green bar (an example is shown). A deletion is presented as a red bar (none is visible on the figure)
Upon zoom-in, SNVs are drawn as pie charts, representing the proportion of each base call at that locus (Figure 5)
Figure 5. Variants track at high power magnification: each SNV is presented as a pie chart and each slice symbolises the relative frequency of each base call (an example is shown). Base call colour codes are given by the track name
At higher modification, insertions are seen as green boxes, with individual inserted bases presented using a pie chart, while deletions look like red boxes and the affected bases are also presented by a pie (Figure 6).
Insertion
Deletion
Figure 6. Variants track at high power magnification: insertion is presented as a green box, deletion is presented as a red box. An example is shown.
Amino Acids Track
Amino acids track becomes available in the Select tracks dialog after completing the Annotate variants task. The actual appearance of the track depends on the zoom level. With low-power magnification, you will see a message View not available at this zoom level, Please zoom in to view amino acids.
When you zoom closer to the genome, all the amino acids become visible as colored boxes (Figure 7) and labeled using the single-letter amino acid code. Alternative amino acids are depicted as additional box on the top of the consensus sequence.
Figure 7. Amino acids track at high power magnification: consensus amino acid sequence is at the bottom of the track, while a variant is shown on the top (change from Threonine to Proline is shown)
If an amino acid spans two exons, its box will be truncated and the line connecting the exons will be dashed. An example is in Figure 8.
Figure 8. Amino acids track: exon-spanning amino acids indicated by truncated boxes (i.e. Alanine on the left) (an example is shown)
An empty gray box on the top of consensus sequence is used to indicate a STOP codon, which is a consequence of a mutation (Figure 9).
Figure 9. Amino acids track: A variant which is in fact a STOP codon is represented by an empty box, as seen on the top of the G (an example is shown)
Untranslated bases, such as ones downstream of a STOP codon are depicted by lighter shades. Figure 10 shows two transcripts in an amino acid track; the direction is from left to right, so amino acids downstream of a STOP codon (P > G > L) are lightly shaded.
Figure 10. Amino acids track: amino acids downstream of a STOP codon are depicted by lighter shades. STOP codon is represented by "." in the middle, direction is from right to left (an example is shown)
Reads Pileup Track
Reads pileup track plots the short sequencing reads, as present in the .bam file. The track is not on by default (go to Select tracks to turn it on) and its appearance depends on the magnification; if you are zoomed out a message - Zoom in to view individual reads - will be displayed.
Forward strand reads are in sky blue, while reverse strand reads are in parakeet green. If paired-end chemistry was used, the paired reads will be depicted as half reads within a gray rectangle encompassing the pair (Figure 11). Singletons will be depicted as thicker reads.
Figure 11. Reads pileup track: short sequencing reads are represented as bars. Paired-end reads are located within a gray box encompassing both pairs. Singletons, such as that on the top right, are depicted as thicker reads (an example is shown)
If you used a junction-aware aligner (such as TopHat or STAR), the junction reads will be depicted using dashed lines, which connect exon-spanning parts of the same read (Figure 12).
Figure 12. Reads pileup track: junction reads are depicted using dashed lines. A RefSeq track is added at the top, to visualise the exons (an example is shown)
Deleted bases can also be seen on a Reads pileup track, as fat black lines (Figure 13).
Figure 13. Reads pileup track: deleted bases depicted using fat black lines (an example is shown)
Probe Intensities Track
Microarray probes are visualised by the Probe intensities track. The probes are shown as bars and their colour depends on the probe intensity, ranging from white (low) to admiral blue (high) (Figure 14).
Figure 14. Probe intensities track: probes are depicted as bars and their colour reflects the intensity (an example is shown)
As with the Reads pileup track, probes may not be visible with low power magnification and you will see a message - Zoom in to view individual microarray probes.
Additional Assistance
If you need additional assistance, please visit our support page to submit a help ticket or find phone numbers for regional support.
Overview
Content Tools