Similarity matrix task is only available on bulk RNA-seq count matrix data node. It is used to compute the correlation of every sample/or feature vs every other sample/or feature. The result is a matrix with the same set of samples/or features on rows and columns, the value in the matrix is correlation coefficient --r.
Click on Similarity matrix task in Correlation section on the menu (Figure 1)
 Figure 1. Similarity matrix menu
When the dialog opens, you will be asked to select whether the calculation samples or features (Figure 2). The are three correlation method options:
Figure 1. Similarity matrix menu
When the dialog opens, you will be asked to select whether the calculation samples or features (Figure 2). The are three correlation method options:
Pearson: linear correlation: 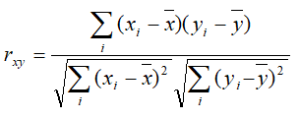
Spearman: rank correlation: 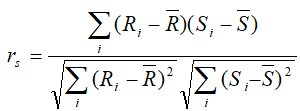
Kendal: rank correlation: 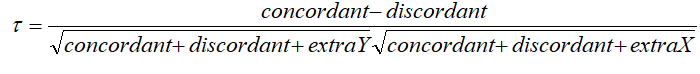
 Figure 2. Similarity matrix dialog
Figure 2. Similarity matrix dialog
An example of the resulting scatterplot is in Figure 3. Each dot is a feature (gene/transcript) while the expression values in the two samples can be read off the coordinate axes, in the same units as present in the data node. For instance, if you normalized your RNA-seq data by transcripts per million (TPM), the coordinate axis will give you expression in TPMs. Pearson’s correlation coefficient and the slope of the regression line are in the upper left corner of the plot.
 Figure 3. Sample correlation plot. Each dot is a sample, the line is the regression line of sample shown on the y-axis over the sample shown on the x-axis. The plot is based on the same expression units as the data that the plot was invoked on
Figure 3. Sample correlation plot. Each dot is a sample, the line is the regression line of sample shown on the y-axis over the sample shown on the x-axis. The plot is based on the same expression units as the data that the plot was invoked on
To visualize a different pair of samples, select another sample from the X axis or Y axis list on the left and push Apply.
Additional Assistance
If you need additional assistance, please visit our support page to submit a help ticket or find phone numbers for regional support.


| Your Rating: |
|
Results: |

|
0 | rates |
Overview
Content Tools