The copy number detection task is used to detect regions of DNA copy number imbalance within the genome for DNA-Seq experiments. Partek Flow provides the CNVkit1 methodology (https://cnvkit.readthedocs.io/en/stable/) to find regions of altered copy number, optimized for targeted resequencing of whole-exome and targeted panels that utilize a hybrid capture approach. The methodology uses both targeted reads and nonspecific off-target reads divided into bins to determine copy number, subsequently normalizing the data to a pooled reference of control samples and correcting for systematic biases.
CNVkit dialog
The CNVkit task can be found under the Copy number analysis tab in the context-sensitive menu when any Aligned reads or Filtered reads data node is selected. The dialog consists of four sections: Define controls, Select Reference Assembly, Target, and Advanced options (Figure 1).
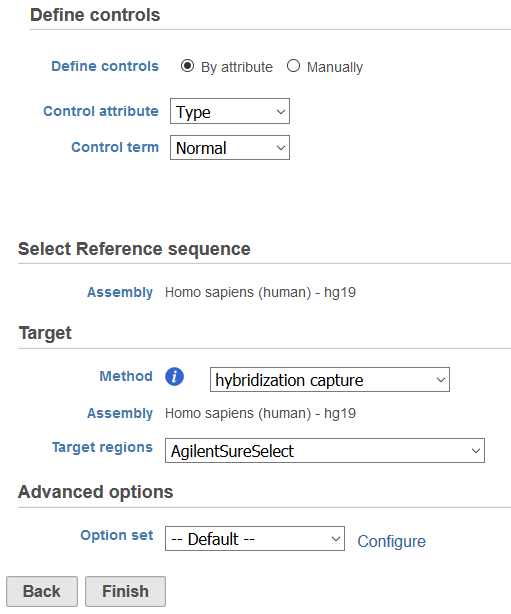
Figure 1. Example of the CNVkit dialog for copy number detection. The projects contains a sample attribute that specifies control.
Define controls allows for the specification of control samples in the project that will be pooled to create a reference copy number for both on- and off-target genomic bins using bias-corrected read depth from each control sample. In projects that contain matched tumor/normal samples, all normal samples should be included in the control sample pool. Control samples can be group based on a categorical attribute in the data tab or manually selected. If no control samples are available, it is possible to run CNVKit with no controls by leaving the sample pool empty. This will create a "flat" reference for neutral copy number.
Select reference sequence will utilize the species genome build utilized for alignment. If the selected aligned data node was imported, the reference assembly used during data alignment needs to be specified from the drop-down list. The Assembly can be previously associated with Partek® Flow® via Library File Management or added on the fly.
The Target section allows for the selection of Method, where the type of sequencing experiment can by specified. While CNVkit is optimized for copy number detection in hybrid capture experiments, it can also analyze whole genome and amplicon sequencing data. Assembly should match the reference sequence previously specified. For hybrid capture or amplicon experiments, Target regions should relate to the regions sequenced in this study using a Gene/feature annotation file. For whole genome data, Annotation will specify a Gene/feature annotation file that will be used to annotate regions of copy number imbalance. The Gene/feature annotation can be previously associated with Partek® Flow® via Library File Management or added on the fly.
Advanced options allows for customization of parameters for CNV detection. Please refer to the CNVkit documentation (https://cnvkit.readthedocs.io/en/stable/) for details on the available parameters.
CNVkit report
Selecting the Task report for the Copy number data node will provide a table with test samples in the project (non-control) and the number of copy number regions associated with each sample (Figure 2). Selecting Option columns at the top right of the table will allow for attribute columns to be added to the report.
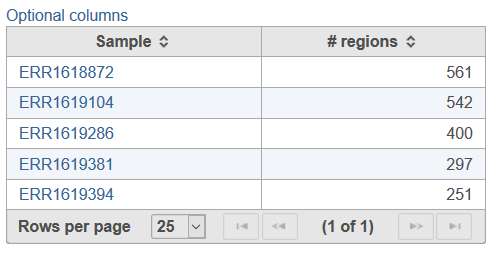
Figure 2. Example of the summary report for CNVkit, providing information on all test samples in the project.
Selecting a sample in the table will open the sample-specific CNVkit report (Figure 3). The sample table will provide a row for each detected region of copy number imbalance in the data with Chromosome, Start and End coordinates. The table also includes the following information:
- gene: content in the copy number region as defined by the associated Target regions or Annotation file
- log2: the weighted log2 mean coverage depth across all bins in the segment
- depth: the weighted mean of absolute-scale mean coverage depth for bins in the segment
- probes: the number of bins covered by the segment
- weight: the sum of bin level weights for the segment that denotes reliability based upon bin size and the square of the log2 spread in the pooled reference
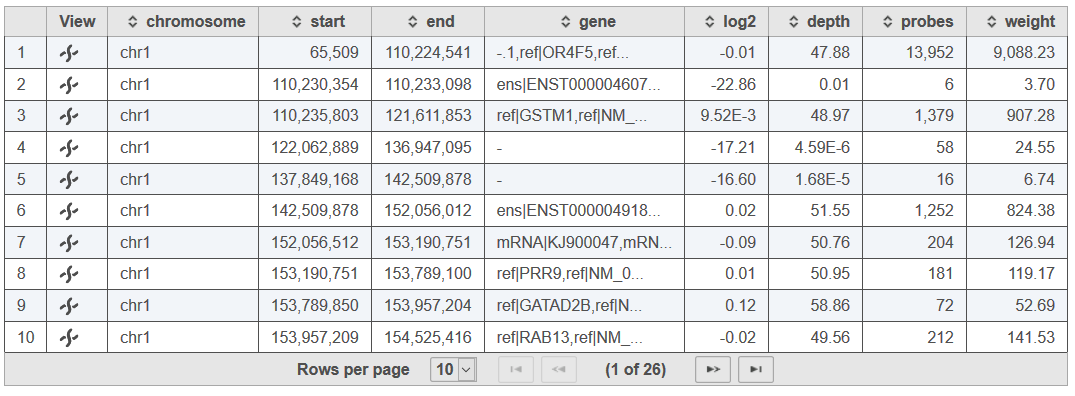
Figure 3. Example of the sample-specific table of regions identified by CNVkit copy number analysis.
Selecting the chromosome icon ![]() in the view column will link to Chromosome view.
in the view column will link to Chromosome view.
References
- Talevich E, Shain AH, Botton T, Bastian BC. CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLOS Comput Biol. 2016;12(4):e1004873. doi:10.1371/journal.pcbi.1004873
Additional Assistance
If you need additional assistance, please visit our support page to submit a help ticket or find phone numbers for regional support.


| Your Rating: |
|
Results: |

|
28 | rates |
Overview
Content Tools