Importing a region list
A region list must contain the chromosome, start location, and stop locations as the first three columns. The chromosome number in the region list must be compatible with the genomic annotation for the species if you plan to use any feature (like motif detection) that requires reference sequence information.
- Import the region list as described above for text files with the following options
- Select Other for data type
- Set chromosome as a text field
- Set location start and stop as either integer or text fields
- Right-click on the imported spreadsheet in the spreadsheet tree
- Select Properties
- Select List of genomic regions from the Configure Spreadsheet dialog to add region to the properties (Figure 1)
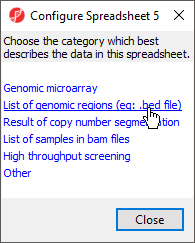
Figure 1. Adding region to the properties of a spreadsheet
The spreadsheet properties will now include region. Alternatively, region can be added as a spreadsheet property from the Configure Genomic Properties dialog by selecting Advanced.. , choosing region from the drop-down menu, selecting Add, and selecting OK.
If you would like to do any operation that requires looking up the reference genomic sequence information for the regions based on genomic location, you will need to specify the species for this region list.
- Right-click on the imported spreadsheet in the spreadsheet tree
- Select Properties
- Select species from the Add Property drop-down menu and click Add
- Specify the Species Name and Genome Build from the drop-down menus
- Select OK
Motif detection
Starting with a region list, you may detect either known or de novo motifs using the ChIP-Seq workflow if your spreadsheet has been associated with a species and a reference genome.
- Select ChIP-Seq from the Workflows drop-down menu
- Select Motif detection from the Peak Analysis section of the workflow
Both Discover de novo motifs and Search for known motifs can be performed. Motif detection sequence information of the genome, you can specify either .2bit file or .fa file which can be used to create .2bit file
Determining the average values for a region list
If you have a region list or a .BED file and you have a microarray experiment with data, you can summarize the microarray data by the genomic coordinates contained in the region list. For example, the region list contains a list of CpG islands, the experiment contains methylation percentage values for probes (β values), and you would like to summarize the methylation values of all probes in each CpG island.
- Import the region list (or .BED file)
Be sure that you have added the region property. The list of region coordinates (chromosome, start, stop) from the region list will be mapped against the reference genome specified for the microarray data so specifying Species and Genome Build for your region list is unnecessary.
- Open the microarray data spreadsheet, this spreadsheet should have annotation file associated to, and there are genomic location information in the annotation file.
Samples should be on rows and data on columns in the microarray data spreadsheet.
- Select the region list spreadsheet
- Right-click any column header in the region list spreadsheet
- Select Insert Average from the pop-up menu (Figure 2)
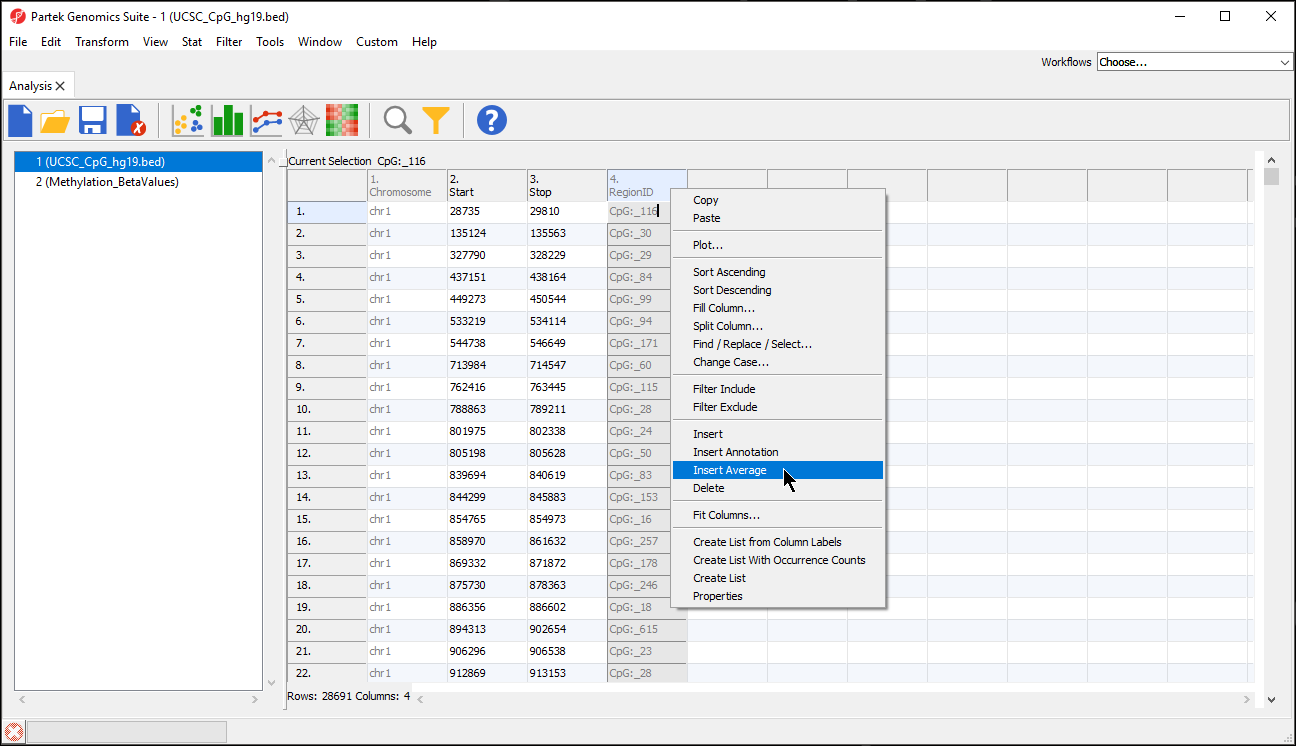
Figure 2. Adding the average values for a region list
- Select the microarray data spreadsheet containing the values you want to average for each region from the Get average from spreadsheet drop-down menu
There are three options for averaging the data (Figure 3). Mean of samples significant in region is used when the region list has SampleIDs from the microarray data set associated with each region. In this case, only the microarray data set samples specified for each region would be included in the mean calculation. Mean of all samples will add columns for the mean value of all probes for all samples and the number of probes for all samples in each region. Mean value for all samples separately will add two columns for each sample with the mean value of all probes for that sample and the number of probes for that sample in each region.
- We have selected Mean value for all samples
- Select OK (Figure 3)
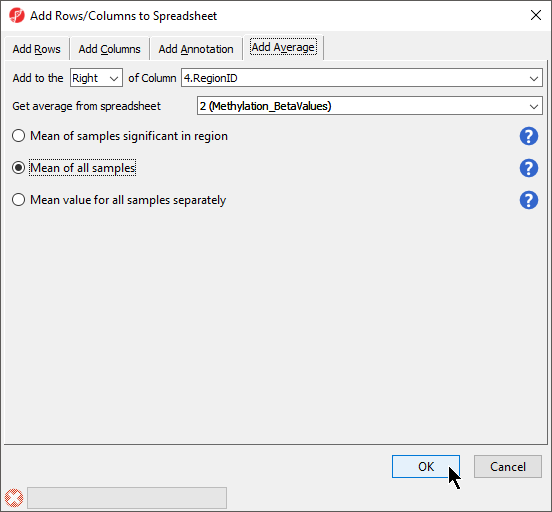
Figure 3. Selecting options for adding average values for regions
Columns will be added to the regions list spreadsheet. Here, we have added two columns with the average β-value for all samples in each CpG island and the number of probes in each CpG island (Figure 4).
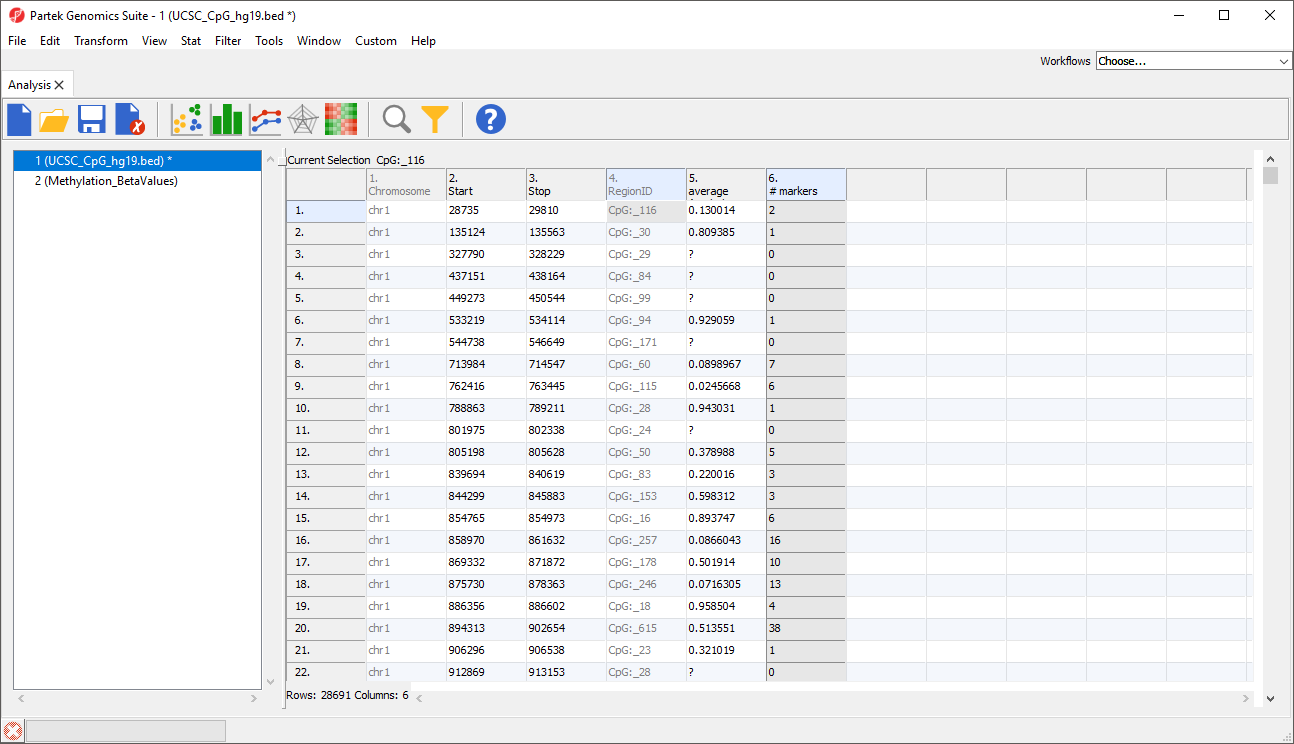
Figure 4. Added average beta values and number of probes per CpG island
Find region overlaps
If you have two or more region lists with coordinates on the same reference genome, you can compare them to identify overlapping regions.
- Open all region list spreadsheets that you want to compare
- Select Tools from the main toolbar
- Select Find Region Overlaps (Figure 5)
Figure 5. Selecting Find Region Overlaps
The Find Region Overlaps tool has two modes of operation. The first, Report all regions, creates a new spreadsheet with any regions that did not intersect and all regions of intersection between any of the input lists. For each intersection, the start and stop coordinates of the intersection and the percent overlap between the intersected region with each of the regions in the input lists are reported. The second, Only report regions present in all lists creates a new spreadsheet with the intersected regions found in all the lists.
- Select your preferred mode; we have selected Only report regions present in all lists
- Select Add New Spreadsheet to add any spreadsheets you want to compare; we are comparing two region list spreadsheets (Figure 6)
- Select OK
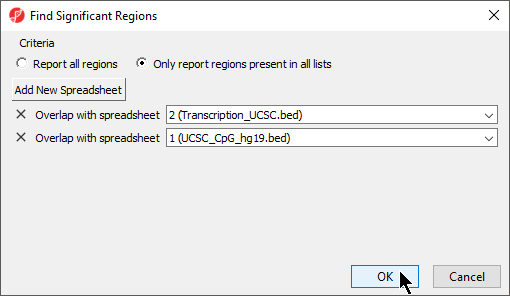
Figure 6. Configuring Find Overlapping Regions
A new region list spreadsheet will be created (Figure 7). The new region list is a temporary spreadsheet so be sure to save it if you want to keep it.
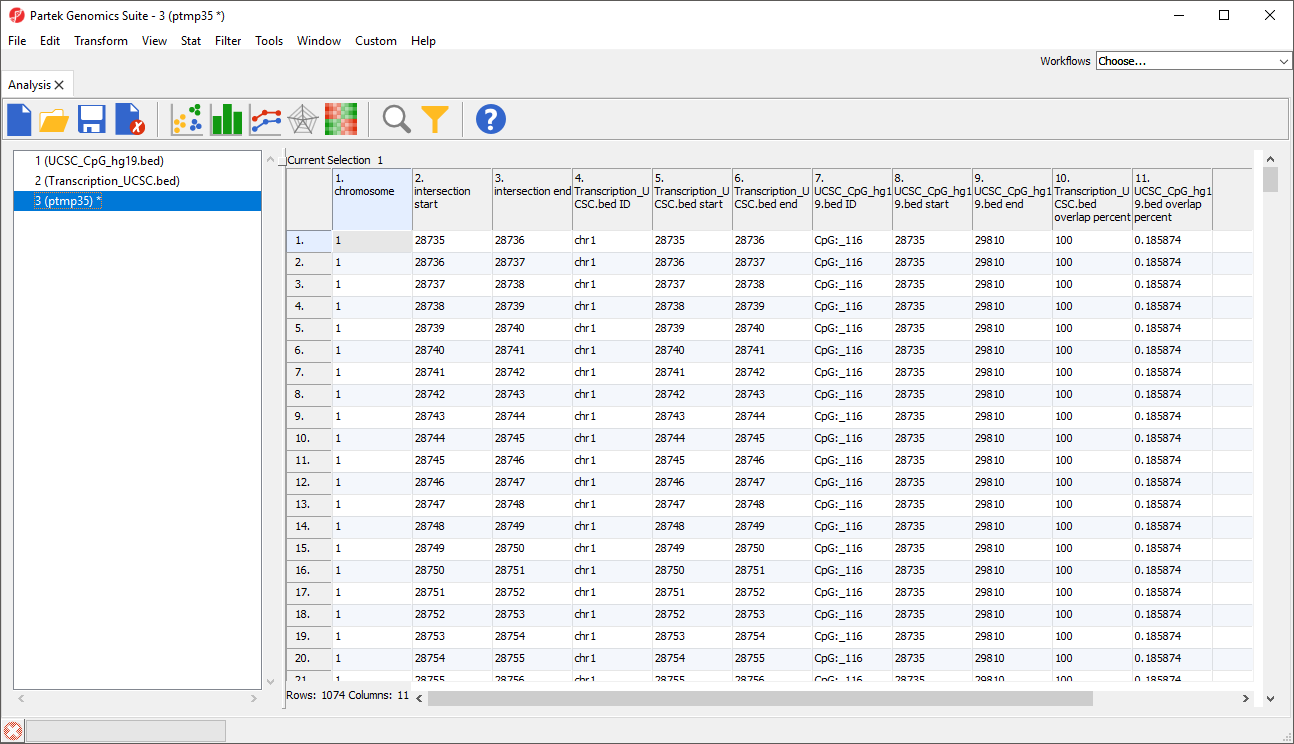
Figure 7. Spreadsheet with regions present in all lists
Importing a genomic position list for SNV annotation
To be annotated using the Annotate SNVs tool, an imported SNV position list must have four columns per locus:
- Position of the SNP listed as chr.basePosition
- Sample ID or name
- The reference base
- The SNP call (sample genotype base)
- Prepare input list as shown (Figure 8) with four columns describing the position, sample, reference base, and sample genotype base for each SNV
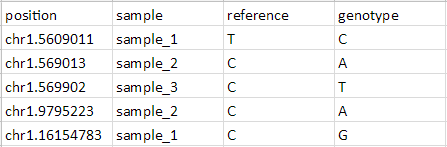
Figure 8. An imported SNV list must follow this format to be annotated by the Annotate SNV tool. The first column must be the position and the position must follow the format shown, chr.basePosition
Save as either a tab-separated or comma separated file
Import the table as a text file
Select Genomic data for What type of data is this file?
Set the position column Type to text
Set the other columns Type to categorical
Select Genomic location instead of marker IDs from the Choose the type of genomic data drop-down menu of the Configure Genomic Properties dialog
Specify the Species and Genome Build
Select OK
The Annotate SNVs tool can now be invoked on this spreadsheet to generate an annotation spreadsheet (Figure 9).
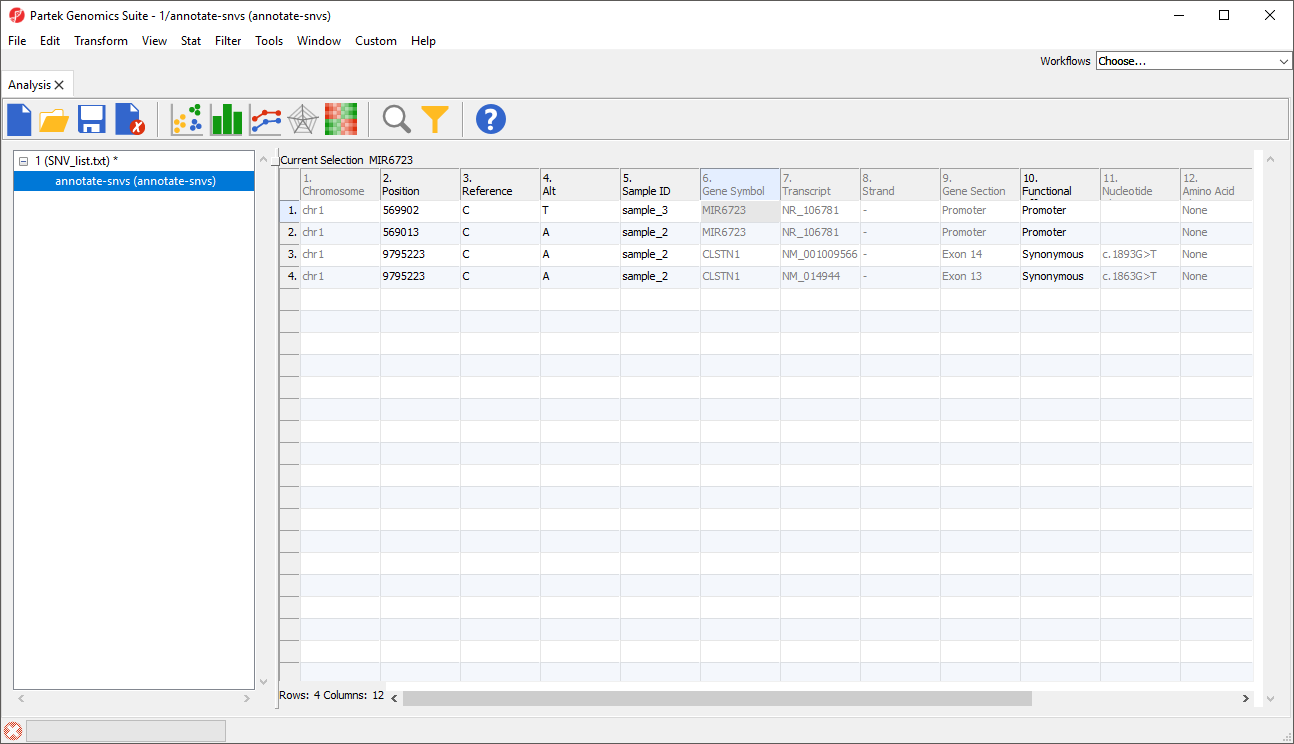
Figure 9. Annotate SNVs creates a new spreadsheet annotating each SNV from the source list
Additional Assistance
If you need additional assistance, please visit our support page to submit a help ticket or find phone numbers for regional support.


| Your Rating: |
|
Results: |

|
35 | rates |
Overview
Content Tools