Page History
...
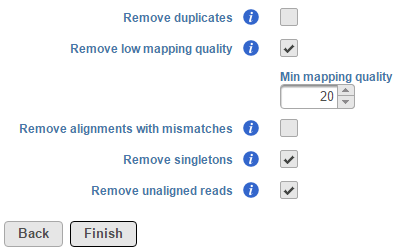
The Filter alignments task can be used to filter aligned reads data using specified parameters. To invoke the task, click on an Aligned reads data node and select Filter alignments. By default, this task removes low-quality reads, singletons and unaligned read information stored within the BAM/SAM file (Figure 1).
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
...
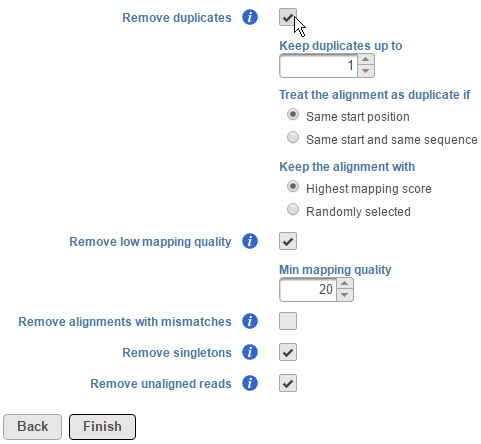
Users also have the option to remove duplicate reads in aligned data. For DNA-Seq analysis, this is typically performed to minimize redundant variant calling information. To remove duplicates, click on the Remove duplicates checkbox (Figure 2).
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
...
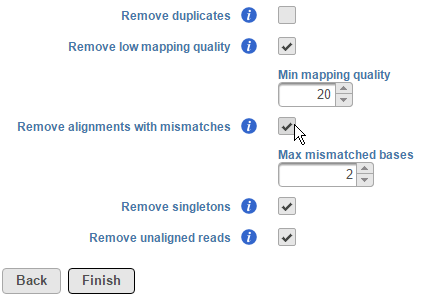
To remove alignments with mismatches, select the Remove alignments with mismatches check box. Using the selector, specify the number the number of mismatched bases that need to be exceeded for the alignment to be excluded (Figure 3). Note that mismatches also include insertions and deletions.
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
...
Overview
Content Tools