Introduction
The Filter alignments task can be used to filter aligned reads data using specified parameters. To invoke the task, click on an Aligned reads data node and select Filter alignments. By default, this task removes low-quality reads, singletons and unaligned read information stored within the BAM/SAM file (Figure 1).
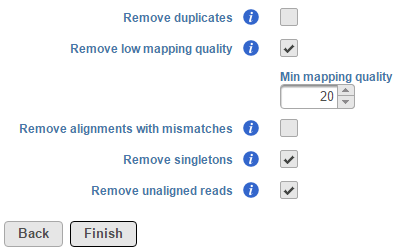 Figure 1. Default filter alignments settings
Figure 1. Default filter alignments settings
Removing duplicates
Users also have the option to remove duplicate reads in aligned data. For DNA-Seq analysis, this is typically performed to minimize redundant variant calling information. To remove duplicates, click on the Remove duplicates checkbox (Figure 2).
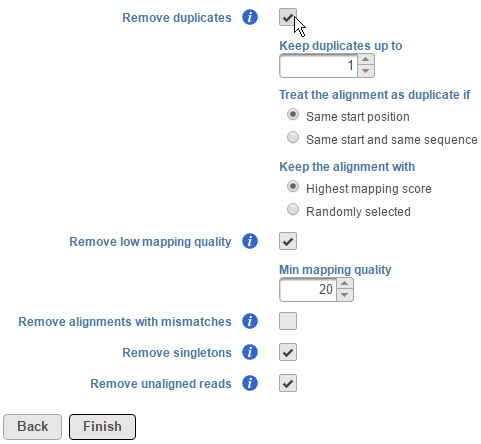 Figure 2. Removing duplicate reads
Figure 2. Removing duplicate reads
Select the number of reads you want to keep. Then specify when alignments are treated as duplicates. This can either be reads that map to the same start position or, additionally, have the same sequence. You can also select whether to keep the read with the highest mapping score or a randomly-selected duplicate.
Remove alignments with mismatches
To remove alignments with mismatches, select the Remove alignments with mismatches check box. Using the selector, specify the number the number of mismatched bases that need to be exceeded for the alignment to be excluded (Figure 3). Note that mismatches also include insertions and deletions.
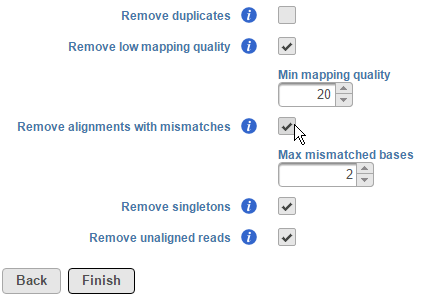 Figure 3. Removing alignments with mismatches
Figure 3. Removing alignments with mismatches
Additional Assistance
If you need additional assistance, please visit our support page to submit a help ticket or find phone numbers for regional support.


| Your Rating: |
|
Results: |

|
44 | rates |
Overview
Content Tools
1 Comment
Melissa del Rosario
author: cignacio