Page History
...
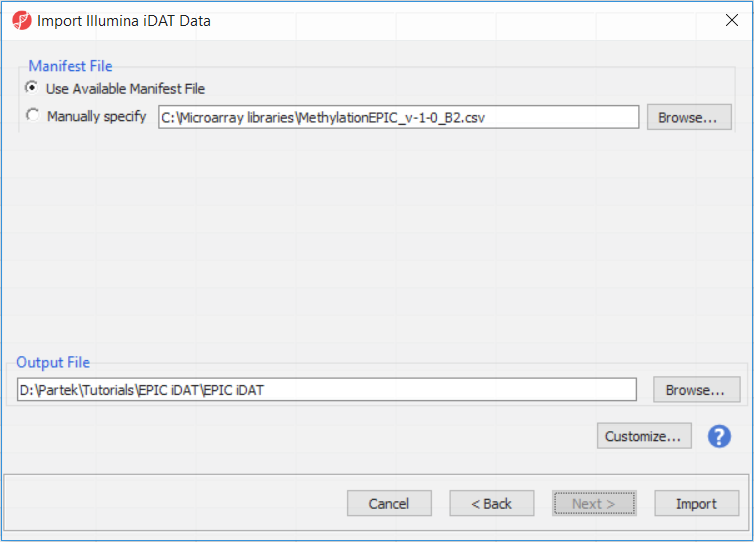
The following dialog (Figure 3) deals with the manifest file, i.e. probe annotation. If a manifest file is not present locally, it will be downloaded in the Microarray libraries directory automatically. The download will take place in the background, with no particular message on the screen and it may take a few minutes, depending on the internet connection. If you have already downloaded the manifest file, to use it, select Use Available Manifest File option. If you have multiple versions available, you can pick an alternative by selecting Manually specify. The latter scenario corresponds to the situation where you want to reanalyze a data set at a later time. To enforce reproducibility, you may want to use the same version of the manifest file that you used when the data was first processed, rather than downloading an up-to-date version.
To proceed with the tutorial, click Import or, if you wish to know more on what is happening during the import, go over the text below.
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
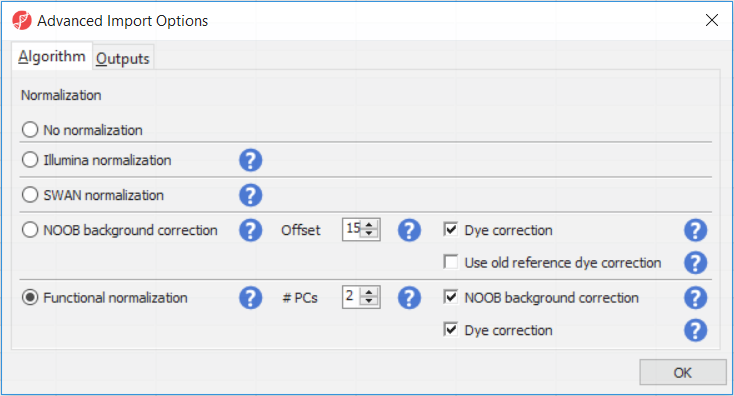
The Customize... button enables a granular control of the import process (Figure 4). First, the normalisation procedure is specified on the Algorithm tab (Figure 4).
Illumina normalisation is the algorithm implemented in GenomeStudio software and provides background subtraction and normalization to control probes. The results in Partek Genomics Suite may differ slightly from the GenomeStudio results, due to differences in selection of the control probes. SWAN normalization stands for subset-quantile within array normalization and was described in the manuscript by Maksimović et al.
Normal-exponential out-of-band (NOOB) background correction corresponds to the Noob background correction method, as implemented in Biocondutor's minfi package. Partek Genomics Suite uses the following options: offset = 15, dyeCorr = TRUE, dyeMethod= ”single” (in the other words, the preprocessFunnorm defaults).
Functional normalisation (default) is an extension of quantile normalisation, that uses control probes to remove the technical variation (Fortin J-P et al., Genome Biol 2014). Partek Genomics Suite uses the same defaults as the minfi package: nPCs = 2, sex = NULL, bgCorr = TRUE, dyeCorr = TRUE. The latter two options refer to the NOOB background correction. PC: principal components.
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
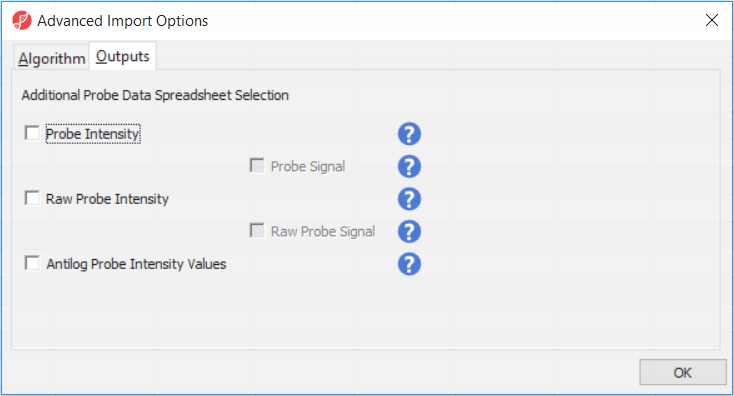
Probe Intensity results in a spreadsheet with samples on rows (with suffix _probe added to the file name), probes on columns, and the sum of raw methylated and unmethylated probe intensities in the cells (the spreadsheet name will also have suffix _probe). Upon selecting Probe Intensity, you can additionally select Probe Signal, which will append rows to the "probe intensity" spreadsheet: for each sample a row with methylated (suffix _meth) and a separate row with unmethylated (suffix _unmeth) intensities (in the other words, three rows per sample).
Raw Probe Intensity results in a spreadsheet with samples on rows (with suffix _raw added to the file name), probes on columns, and the sum of raw red and green probe intensities in the cells (the spreadsheet name will also have suffix _raw). Upon selecting Raw Probe Intensity, you can additionally select Raw Probe Signal, which will append rows to the "raw probe intensity" spreadseet: for each sample a row with red (suffix _red) and a separate row with green (suffix _green) intensities (in the other words, three rows per sample).
All the values discussed above will be log2 transformed upon import, and if you want to see the values in the linear space, select Antilog Probe Intensity Values.
Probe intensities and raw probe intensities can be used for advanced troubleshooting purposes and antiloged probe intensities can be used for copy number detection.
Section Heading
Section headings should use level 2 heading, while the content of the section should use paragraph (which is the default). You can choose the style in the first dropdown in toolbar.
...
Overview
Content Tools