Page History
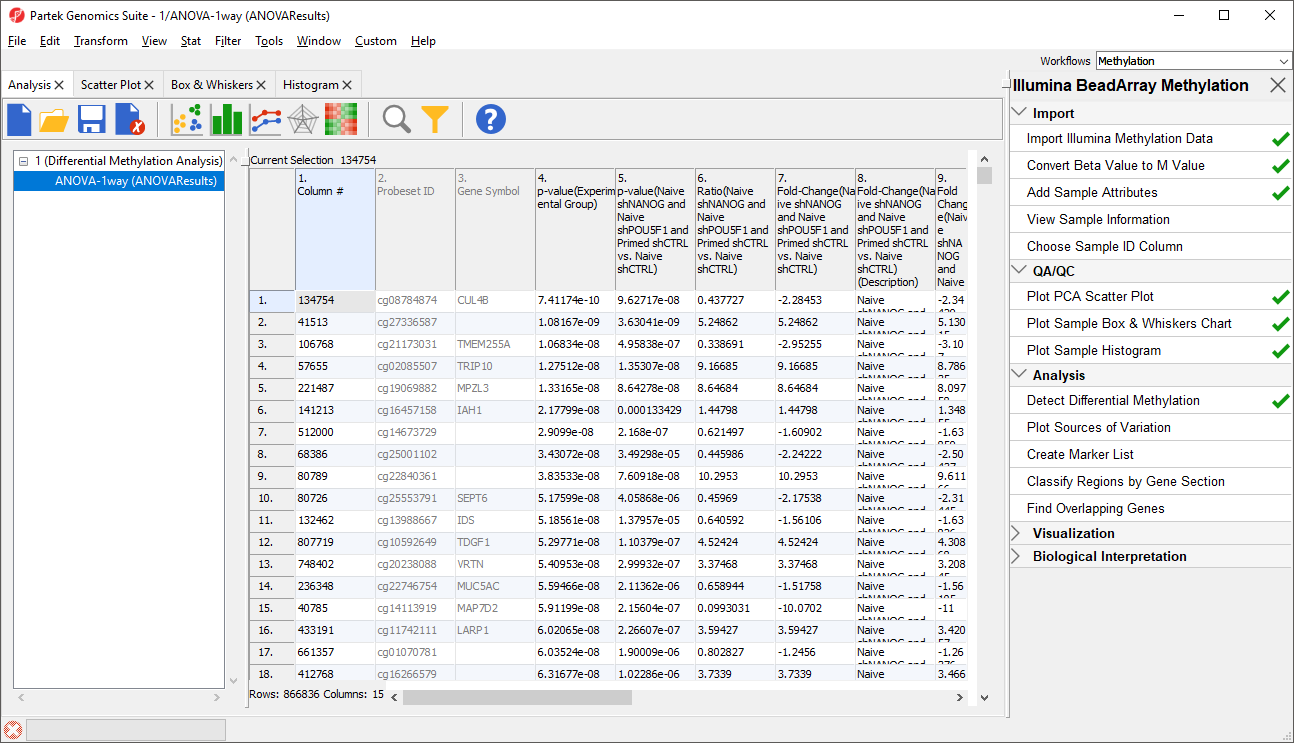
To detect differential methylation between CpG loci in different experimental groups, we can perform an ANOVA test. For this tutorial, we will perform a simple onetwo-way ANOVA to compare the methylation states of the four two experimental groups.
- Select Defect Detect Differential Methylation from the Analysis section of the Illumina BeadArray Methylation workflow
A new child spreadsheet, mvalue, is created when Detect Differential Methylation is selected. M-values are an alternative metric for measuring methylation. β-values can be easily converted to M-values using the following equation: M-value = log2( β / (1 - β)).
An M-value close to 0 for a CpG site indicates a similar intensity between the methylated and unmethylated probes, which means the CpG site is about half-methylated. Positive M-values mean that more molecules are methylated than unmethylated, while negative M-values mean that more molecules are unmethylated than methylated. As discussed by Du and colleagues, the β-value has a more intuitive biological interpretation, but the M-value is more statistically valid for the differential analysis of methylation levels.
Because we are performing differential methylation analysis, Partek Genomics Suite automatically creates an M-values spreadsheet to use for statistical analysis.
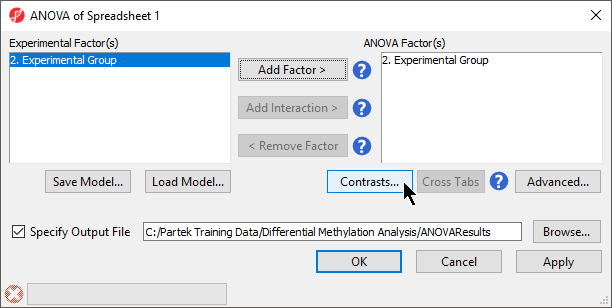
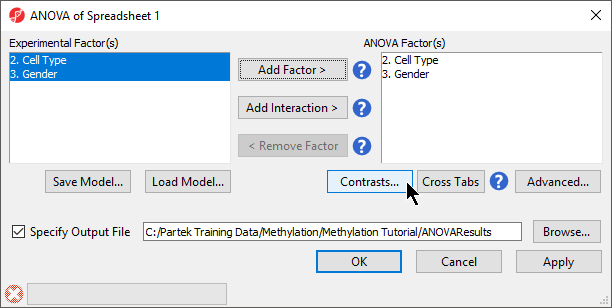
- Select 2. Cell Type and 3. shRNA treatment Gender from the Experimental Factor(s) panel
- Select Add Factor > to move 2. Cell Type and 3. shRNA treatment Gender to the ANOVA Factor(s) panel (Figure 1)
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
- Select Contrasts...
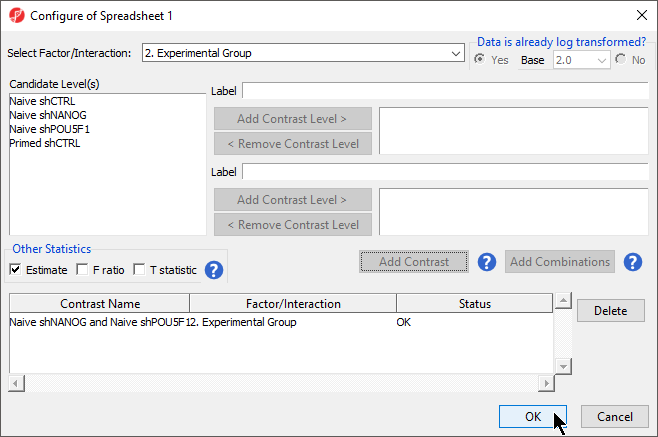
- Select Yes for Leave Data is already log transformed? because M-values are based on logit transformation
- Select Naive shPOU5F1
- Select set to No
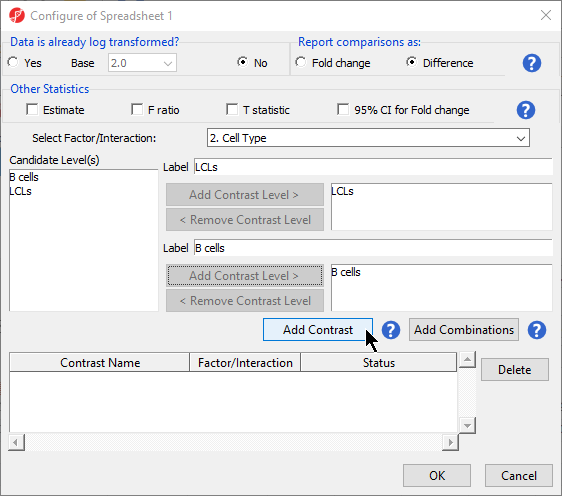
- Leave Report comparisons as set to Difference
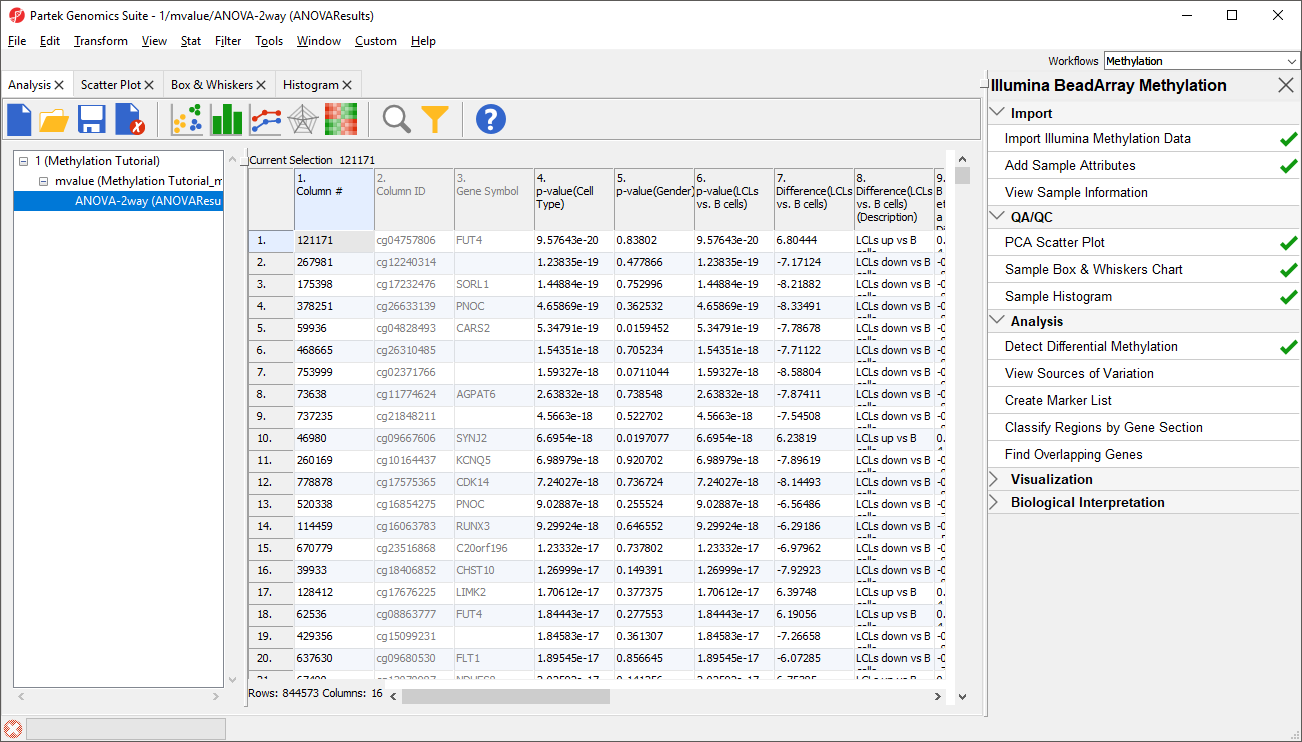
For methylation data, fold-change comparisons are not appropriate. Instead, comparisons should be reported as the difference between groups.
- Select 2. Cell Type from the Select Factor/Interaction drop-down menu
- Select LCLs
- Select Add Contrast Level > for the upper group
- Repeat to add Naive shNANOG and Primed shCTRL to the upper group
- Select Naive shCTRL
- Select Select B cells
- Select Add Contrast Level > for the lower group
- Select Add Contrast Select the Estimate box in the Other Statistics section of the Configure dialog (Figure 2)
By default, the fold-change value for each contrast will be calculated. Selecting Estimate will also include the difference in methylation levels between the groups at each CpG site in the output. These values will be needed later in the tutorial when we filter the differentially methylated loci.
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
- Select Add CombinationSelect OK to close the Configuration dialog
...
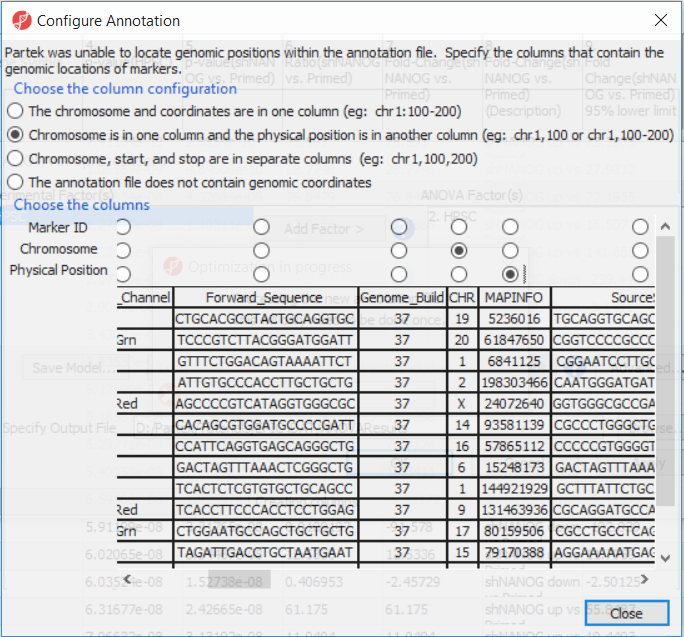
- Select Chromosome is in one column and the physical location is in another column for Choose the column configuration
- Select Ilmn ID for Marker ID
- Select CHR for Chromosome i
- Select MAPINFO for Physical Position
- Select Close
Select Close. This enable Partek enables Partek Genomics Suite to parse out probe annotation annotations from the manifest file.
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
|
| Page Turner | ||
|---|---|---|
|
...
Overview
Content Tools