Page History
When the reads are aligned to a genome reference, e.g. hg38, the quantification is performed on transcriptome, you need to provide the annotation model file of the transcriptome.
Quantification dialog
If the alignment was generated in Partek® Flow®, the genome assembly will be displayed as text on the top of the page In ChIP-seq or ATAC-seq analysis, after peak regions detected, quantify regions will report the read count from the peaks for each sample. The quantify regions algorithm is the same as Quantify to annotation model (Partek E/M), the annotation model is location of the peaks.
Quantify regions dialog
Click on peak data node, choose Quantify regions task in Quantification section. (Figure 1), you do not have the option to change the reference.
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|

need to specify annotation model since the regions used to generate read counts is a list of peak locations from all the samples generated in the peak detection task.
If the bam file is imported, you need to select the assembly with which the reads were aligned to, and which annotation model file you will use to quantify from the drop-down menus (Figure 2).
...
| Numbered figure captions | ||||||
|---|---|---|---|---|---|---|
|
| |||||
|

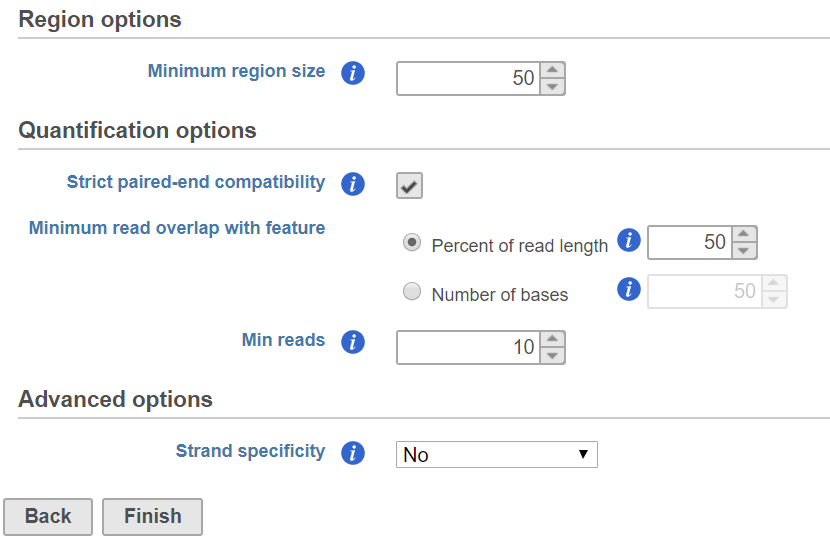
In the Quantification options section, when the Strict paired-end compatibility check button is selected, paired end reads will be considered compatible with a transcript only if both ends are compatible with the transcript. If it is not selected, reads with only one end have alignment that is compatible with the transcript will also be counted for the transcript .
...
Overview
Content Tools