In ChIP-seq or ATAC-seq analysis, a major challenge after detecting enriched regions or peaks is to compare samples and identify differentially enriched regions. In order to compare samples, a common set of regions must be identified and the number of reads mapping to each region quantified. The Quantify regions task addresses this challenge by generating a union set of unique regions and reporting the number of reads from each sample mapping to each region.
To run Quantify regions:
- Click a Peaks data node
- Click the Quantification section in the toolbox
- Click Quantify regions
Quantify regions method
The Quantify regions task takes MACS2 output, a Peaks or Annotated Peaks data node, as its input. In a typical ATAC-Seq or ChIP-Seq analysis, MACS2 is configured to output a set of enriched regions or peaks for each experimental sample or group individually. Quantify regions takes these sets of regions and merges them into a union set of unique regions that it saves as a .bed file. To combine the region sets, overlapping regions between samples/groups are merged. Where overlap ends, a break point is created and a new region defined. All non-overlapping or unique regions from each sample/group are also included.
For example, consider an experiment where MACS2 detected enriched regions for two samples, Sample A and Sample B. In Sample A, a region is detected on chromosome 1 from 100bp to 300bp, chr1:100-300. In Sample B, a region is detected at chr1:160-360. The Quantify regions task will give the following union set of unique regions for these partially overlapping regions:
chr1:100-160 (region detected in Sample A only)
chr1:160-300 (region detected in both Sample A and Sample B)
chr1:300-360 (region detected in Sample B only)
After generating a .bed file with the union set of unique regions, Quantify regions performs quantification using the same algorithm as Quantify to annotation model (Partek E/M) with the .bed file as the annotation model.
Configuring Quantify regions
The Quantify regions dialog includes configuration options for generating the union set of unique regions and quantifying reads to the regions (Figure 1).
When regions from multiple samples are combined, a small offset in position between enriched regions in different samples can result in many very short unique regions in the union set. The Minimum region size option lets you filter out these very short regions. If a region is smaller than the specified cutoff, the region is excluded. By default, this is set to 50bp, but may need to be adjusted depending on the size of regions you expect to see in your assay.
Quantification options are the same as in the Quantify to annotation model (Partek E/M) dialog. The Percent of read length is set to 50% by default to account for small offsets in position between enriched regions in different samples.
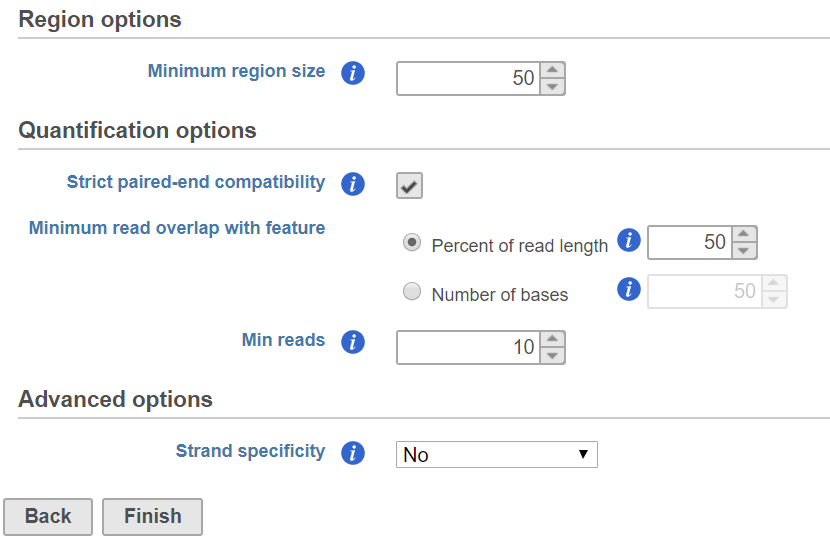
Figure 1. Quantify regions dialog
Quantify regions output
Quantify regions generates a counts data node with the number of counts in each region for each sample. This data node can be annotated with gene information using the Annotate regions task and analyzed using tasks that take counts data as input, such as normalization, PCA, and ANOVA. For ChIP-Seq experiments with input control samples, the Normalize to baseline task can be used prior to downstream analysis.
Similar to the Quantify to annotation model (Partek E/M) task report, the Quantify regions task report includes feature distribution information including a descriptive stats table, a distribution bar chart, a sample box plot, and sample histogram (Figure 2).
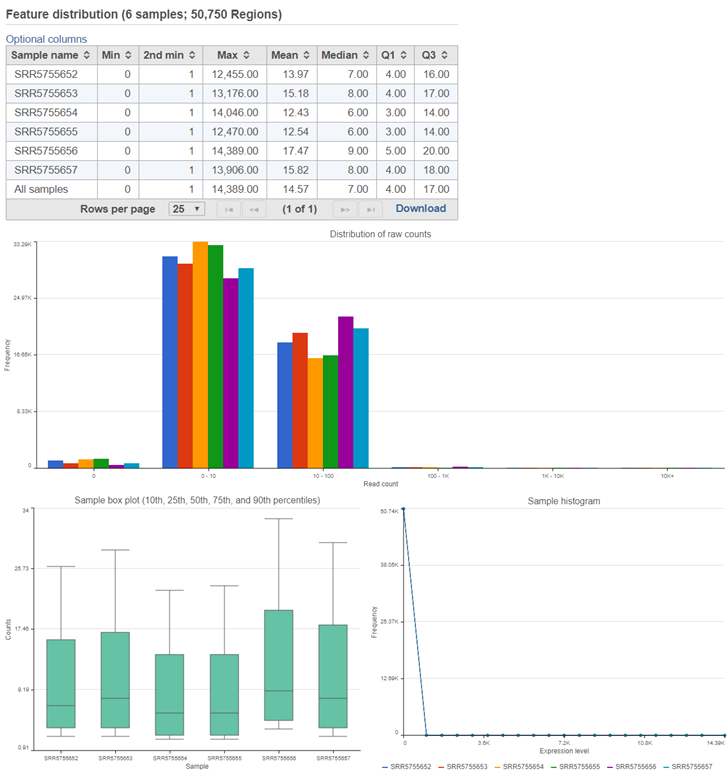
Figure 2. Quantify regions task report
To download the .bed file with the union set of unique regions, click the Quantify regions task node, click Task details, click the regions.bed file in the Output files section, and click Download.
References
- Xing Y, Yu T, Wu YN, Roy M, Kim J, Lee C. An expectation-maximization algorithm for probabilistic reconstructions of full-length isoforms from splice graphs. Nucleic Acids Res. 2006; 34(10):3150-60.
Additional Assistance
If you need additional assistance, please visit our support page to submit a help ticket or find phone numbers for regional support.


| Your Rating: |
|
Results: |

|
28 | rates |
Overview
Content Tools