We are now ready to measure gene expression in our dataset. To do this, we will use the mRNA quantification task in the Analyze Known Genes section of the RNA-Seq workflow. mRNA quantification creates spreadsheets showing expression at exon, transcript, and gene levels and reports raw and normalized reads for each sample.
Please note that the normalization method used by Partek Genomics Suite is Reads Per Kilobase per Million mapped reads (RPKM) (Mortazavi et al. 2008). In brief, this normalization method counts total reads in a sample, divides by one million to create a per million scaling factor for each sample; then divides the read counts for the feature (exon, transcript, or gene) by the per million scaling factor to normalize for sequencing depth and give a reads per million value; and finally divides reads per million values by the length of the feature (exon, transcript, or gene) in kilobases to normalize for feature size.
- Select 1 (RNA-Seq) from the spreadsheet tree
- Select mRNA quantification in the Analyze Known Genes section of the RNA-seq workflow
The RNA-Seq Quantification dialog will appear (Figure 1).
- Select RefSeq Transcripts 2017-05-02 from the mRNA section of the Specify a database of genomic features to quantify panel of the dialog
Your choices in the Configure the test panel of the dialog depend on the design and aims of your experiment. A detailed description of each option can be viewed by selecting the (![]() ) icon next to it.
) icon next to it.
- For Strand-specificity: select No
Your choice here depends on the method used for sample preparation. A directional mRNA-seq sample preparation protocol only synthesizes the first strand of cDNA whereas other methods reverse transcribe the mRNA into double-stranded cDNA. If double-stranded cDNA has been synthesized, the sequencer reads sequences from both the forward and reverse strands but does not discriminate between them, eliminating strand information. When strand information is preserved, it is possible for paired-end sequences to come from a combination of the forward and reverse strands. If in doubt, select Auto-detect from the drop-down list. The data for this tutorial did not preserve strand information so we selected No.
- For In the gene-level result report intronic reads as compatible with the gene?, select No
Selecting Yes would include intronic reads in the gene-level results, which might be useful for discovering unannotated transcripts for known genes, and also includes introns in the RPKM calculation for the gene-level results.
- For Require strict paired-end compatibility select No
Selecting Yes would require that two alignments form the same read must map to the same transcript to be considered compatible. However, the data set used in this tutorial consists of single-end reads so this option is unnecessary.
- For report results with no reads from any sample? select No
Selecting Yes would include all the genes/transcripts/exons in the transcriptome, even if there are no reads for that feature from any sample.
- Make sure Report unexplained regions with more than ___ reads is selected and specify 5 as the number of reads
This option will create a spreadsheet that includes all regions with a specified number of reads that map to the genome, but not to any feature included in the selected database of genomic features.
- Select Report exon-level results
If selected, spreadsheets will be created describing expression at the exon level.
Your RNA-Seq Quantification dialog should now be configured as shown (Figure 1). Descriptions of the spreadsheets that can be created by mRNA Quantification can be viewed by selecting Describe results to bring up the Quantification Result Help dialog.
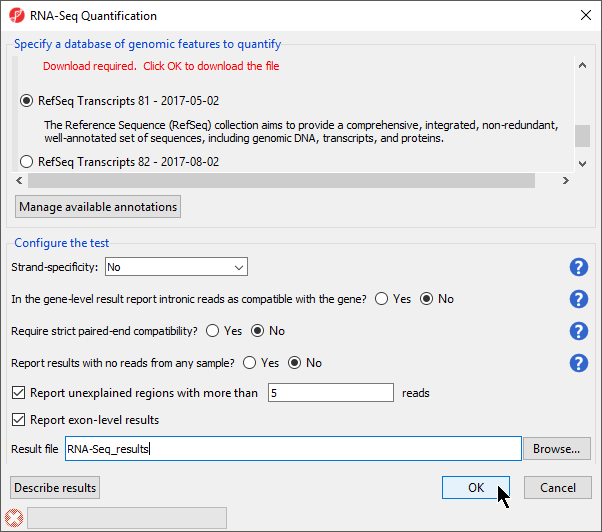
Figure 1. Configuring the RNA-Seq Quantification dialog
- Select OK to perform the RNA-Seq quantification
Reads will now be assigned to individual transcripts of a gene based on the Expectation/Maximization (E/M) algorithm (Xing, et al. 2006). In Partek Genomics Suite software, the E/M algorithm is modified to accept paired-end reads, junction aligned reads, and multiple aligned reads if these are present in your data. For a detailed description of the E/M algorithm, refer to the RNA-Seq white paper (Help > On-line Tutorials > White Papers). Several spreadsheets containing the analyzed results will be generated. Progress bars in the lower left-hand corner RNA-Seq Quantification window and the main window will update as the data is analyzed.
- If you have not disabled it, the the Quantification Result Help dialog will appear. Select Close
The Analysis tab now shows the spreadsheets created by mRNA Quantification in the spreadsheet tree as a child spreadsheet of 1 (RNA-seq) (Figure 2).
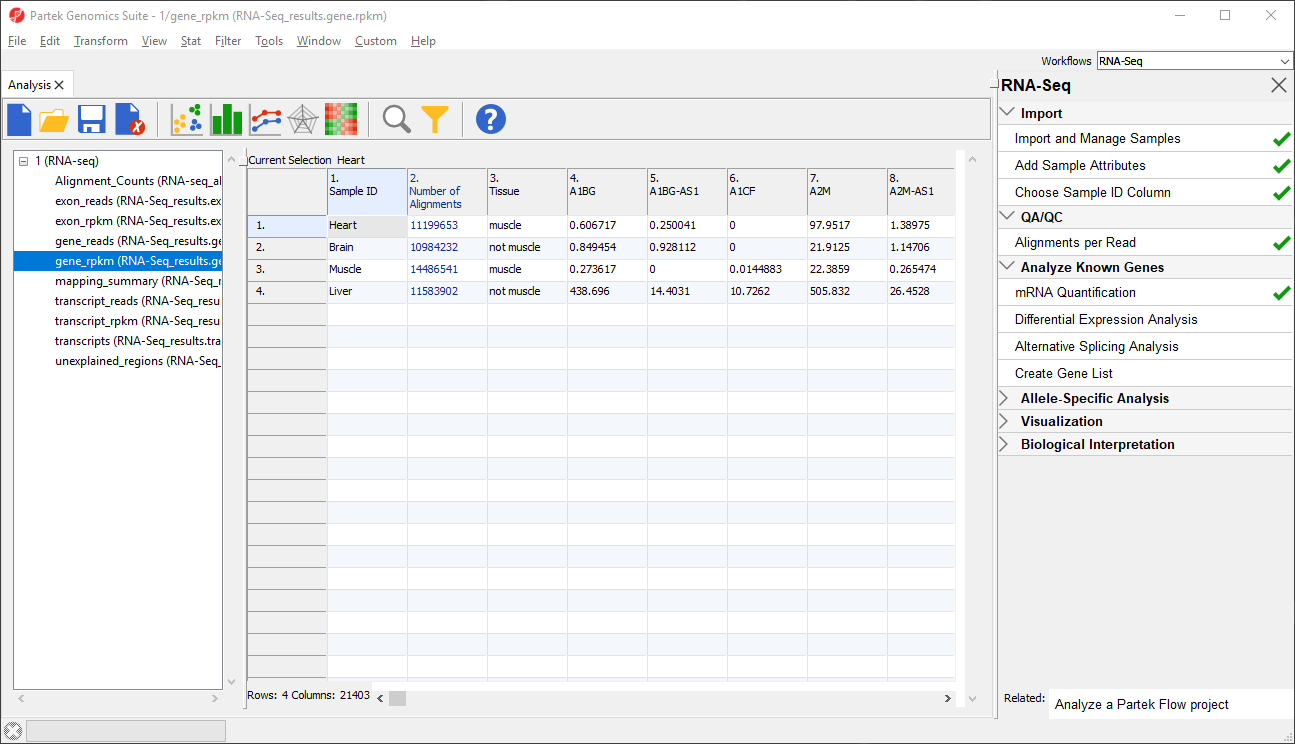
Figure 2. Viewing the results of mRNA Quantification
The _reads and _rpkm spreadsheets
Data on features - genes, transcripts, and exons - are presented before and after normalization as _reads and _rpkm spreadsheets. In this tutorial, we have created exon_reads, exon_rpkm, gene_reads, gene_rpkm, transcript_reads, and transcript_rpkm spreadsheets.In these spreadsheets, samples are listed one per row and the normalized counts of the reads mapped to features are in columns (Figure 2).
The _reads and _rpkm spreadsheets can be used for data analysis. Sample grouping can be visualized using PCA. Select View > Scatter Plot from the toolbar or press ![]() on the quick action bar to create a PCA plot from the selected spreadsheet. See Exploring gene expression data for an example of using PCA plots for data analysis or consult Chapter 7 of the Partek User's Manual for a detailed introduction to PCA. With replicates in a sample group, you would also be able to use the _rpkm spreadsheet to perform differential expression analysis using ANOVA.
on the quick action bar to create a PCA plot from the selected spreadsheet. See Exploring gene expression data for an example of using PCA plots for data analysis or consult Chapter 7 of the Partek User's Manual for a detailed introduction to PCA. With replicates in a sample group, you would also be able to use the _rpkm spreadsheet to perform differential expression analysis using ANOVA.
The transcripts spreadsheet
The transcripts spreadsheet lists a transcript in each row.
It is possible to derive basic information from the RNA-Seq_result.transcripts spreadsheet about differential and alternative splicing between your samples even if you don’t have replicates using a simple chi-squared or log-likelihood tests because each sample is represented only once and we can assume a null hypothesis that the transcripts are evenly distributed across all samples. However, the power of Partek Genomics Suite software resides in the implementation of a mixed-model ANOVA that can handle unbalanced and incomplete datasets, nested designs, numerical and categorical variables, any number of factors, and flexible linear contrasts when you do have biological replicates.
The unexplained_regions spreadsheet
The contents of this spreadsheet are explained in more detail in a later section of the tutorial - Analyzing the unexplained regions spreadsheet.
References
Mortazavi, A., Williams, B.A., McCue, K., Schaeffer, L., & Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature, 2008; 5: 621-8.
Xing Y, Yu T, Wu YN, Roy M, Kim J, Lee C: An expectation-maximization algorithm for probalisitic reconstructions of full-length isoforms from splice graphs. Nucleic Acids Res 2006, 34: 3150-3160.
Additional Assistance
If you need additional assistance, please visit our support page to submit a help ticket or find phone numbers for regional support.


| Your Rating: |
|
Results: |

|
36 | rates |
Overview
Content Tools