miRNAs regulate gene expression at the post-transcriptional level by base-pairing with the three prime untranslated region (3’ UTR) of the target gene, causing cleavage/degradation of the cognate mRNA or preventing translation initiation. Integration of miRNA expression with gene expression data to study the overall network of gene regulation is vital to understanding miRNA function in a given sample. Partek Genomics Suite provides a platform that can analyze miRNA and gene expression data independently, yet allows data to be integrated for downstream analysis.This integrative analysis can be accomplished at several different levels. If you only have miRNA data, then Partek Genomics Suite can search the predicted gene targets in a miRNA-mRNA database like TargetScan to provide a list of genes that might be regulated by the differentially expressed miRNAs. Alternatively, if you have only gene expression data, Partek Genomics Suite can use the same database to identify the microRNAs that putatively regulate those differentially expressed genes in a statistically significant manner. If you have gene expression data and miRNA data from comparable tissue/species, Partek Genomics Suite can combine the results of these separate experiments into one spreadsheet. Lastly, if the miRNA and mRNA from the same source was analyzed (as in this tutorial), then you may statistically correlate the results of miRNA and gene expression assays.
Finding putative genes regulated by miRNAs
This application is useful in the case where you have miRNA expression data, but not gene expression data. Using a database like TargetScan, microCosm, or a custom database, you can identify the list of genes that are predicted to be regulated by these differentially expressed miRNAs and then perform Biological Interpretation tasks on the list of genes.
- Select Combine miRNAs with their mRNA targets from the miRNA Integration section of the miRNA Expression workflow
- Select the Get All Targets tab
- Select TargetScan7.1 for Database Name
- Select brain vs. heart human for Spreadsheet Name
- Set Column with microRNA labels to 2. Probeset ID
- Name the Result file PutativeGenes
- Select OK (Figure 1)
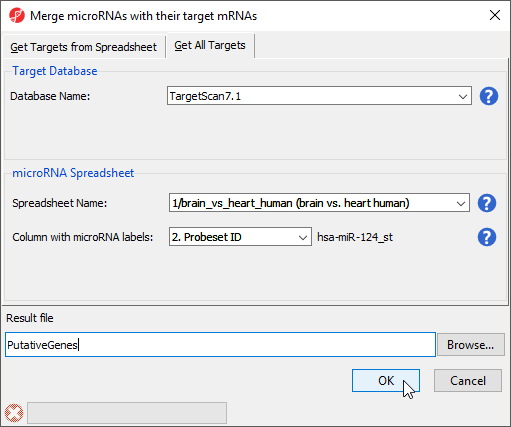
Figure 1. Identifying all predicted gene targets of differentially expressed miRNAs
This will create a new spreadsheet PutativeGenes that contains a miRNA and a putative gene target in each row. Because each miRNA can regulate multiple genes, the list will be much longer than the input miRNA list. Each row contains a gene so this spreadsheet can be analyzed using GO Enrichment and Pathway Enrichment tasks from the Biological Interpretation section of the workflow.
Another useful way to analyze this list is to determine which genes could be targeted by multiple miRNAs in the list. To do this:
- Right-click on the column 13. Gene Symbol header
- Select Create List With Occurrence Counts from the pop-up menu (Figure 2)
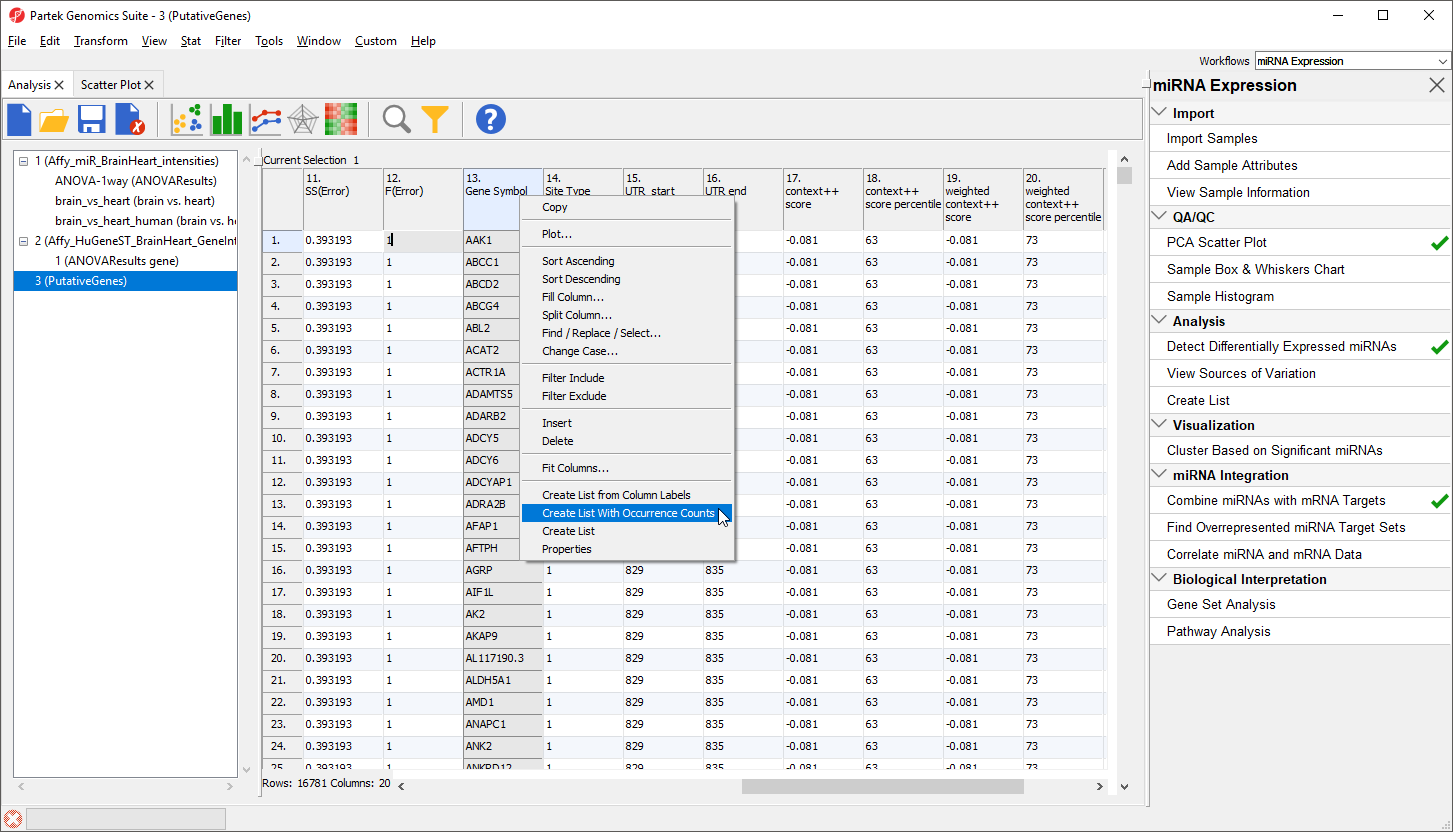
Figure 2. Creating an occurrence counts list from the list of putative miRNA target genes
The new spreadsheet is a temporary spreadsheet listing each gene in alphabetical order and giving the occurance count of each. Sorting by descending order will list the gene with the most occurances first (Figure 3).
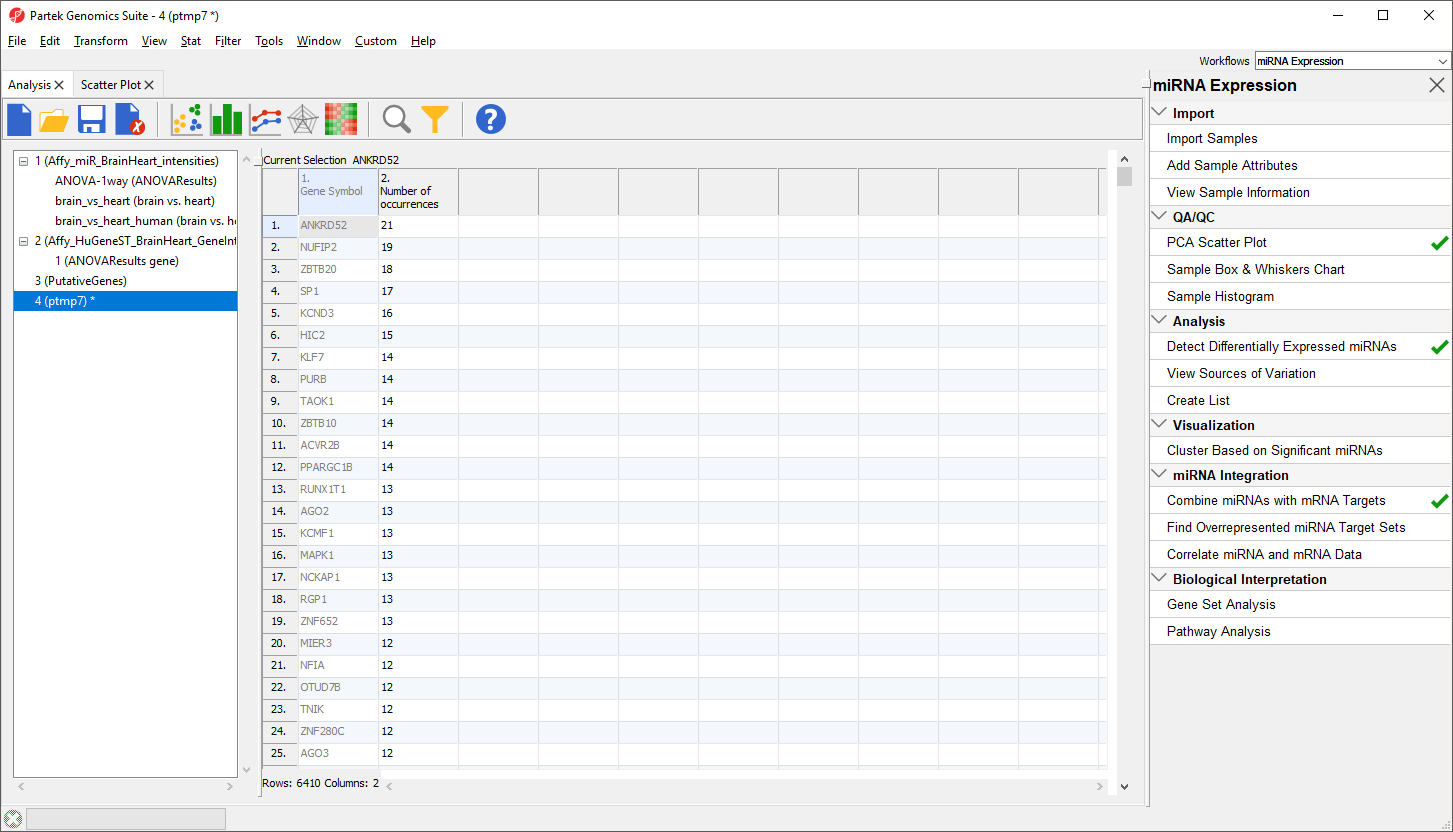
Figure 3. Occurrence list of putative miRNA target genes
We will not be using this temporary spreadsheet moving forward. You can close the spreadsheet by selecting ![]()
Finding overrepresented miRNA targets sets from gene expression data
This application is useful when you only have gene expression results or a gene list of interest and are interested in identifying which miRNAs might regulated the genes. Using a databse like TargetScan, you can create a list of miRNAs that are statistically predicted to regulated those genes. miRNAs of particular interest could then be explored using a lower-throughput technique like RT-qPCR.
Using the gene list as input, a Fisher's Exact right-tailed p-value is calculated to show the overrepresentation of genes of interest for each miRNA in the database. The smaller the p-value, the more overrepresented the miRNAs are for the dataset. Target associations are taken from a database, TargetScan in this example. If the input list is a filtered list of genes from an ANOVA calculation, the parent spreadsheet is used to identify the background list of genes from the array. Genes in the array but not in the significant gene list will be treated as background in the calculations.
To begin, we need to create a list of significant genes using the ANOVAResults gene spreadsheet.
- Select the ANOVAResults gene spreadsheet in the spreadsheet tree
- Select Create List from the workflow
- Select Brain vs. Heart
- Set the Save list as to brain vs. heart genes
- Leave other fields at their default values (Figure 4)
- Select Create
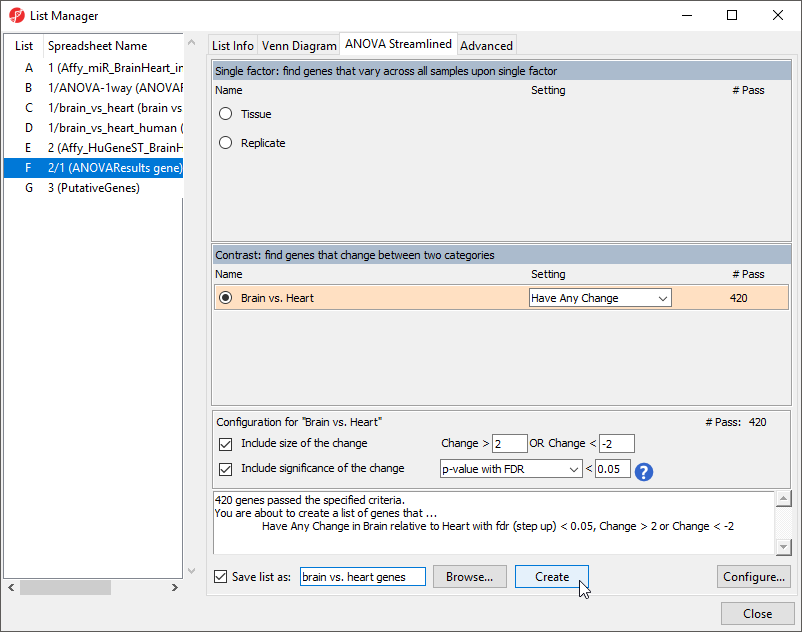
Figure 4. Creating a list of significantly differentially expressed genes
- Select Close to exit the List Manager dialog
We will now use this list to identify overrepresented miRNA target sets.
- Select Find overrepresented miRNA target sets from the miRNA Integration section of the workflow
- Select TargetScan 7.1 from the Target Databse drop-down menu
- Select brain vs. heart genes from the mRNA Spreadsheet drop-down menu
- Select 4. Gene Symbol from the Column with gene symbols drop-down menu (Figure 5)
- Select OK
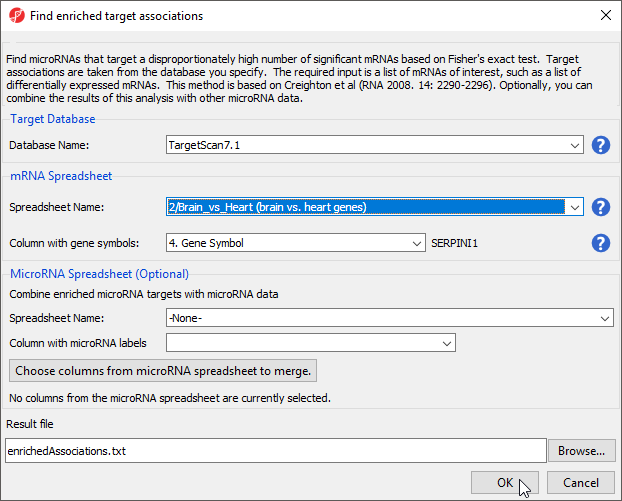
Figure 5. Finding enriched miRNA target sets
A new spreadsheet, enrichedAssociations, will be created with miRNAs from the database on rows (Figure 6). Column 1 contains the miRNA name and column 2 shows its p-value. The smaller the p-value, the more significant it is. Column 3 contains the number of genes from the (input) significant gene list that are targeted by this microRNA and Column 7 shows the number of significant genes from the input list that are not targeted by this microRNA. Columns 4 and 5 contain the number of significantly up- and down-regulated genes from the input significant gene list targeted by the miRNA. Column 6 shows the number of background genes (genes on the array but not in the input significant gene list) that are targeted by the miRNA and Column 8 shows the number of background genes on the array that are not targeted by the miRNA. The numbers in columns 3, 6, 7 and 8 will be used to calculate the Fisher’s Exact (right-tailed) p-value, a measure of the overrepresentation of the predicted miRNAs within a gene set.
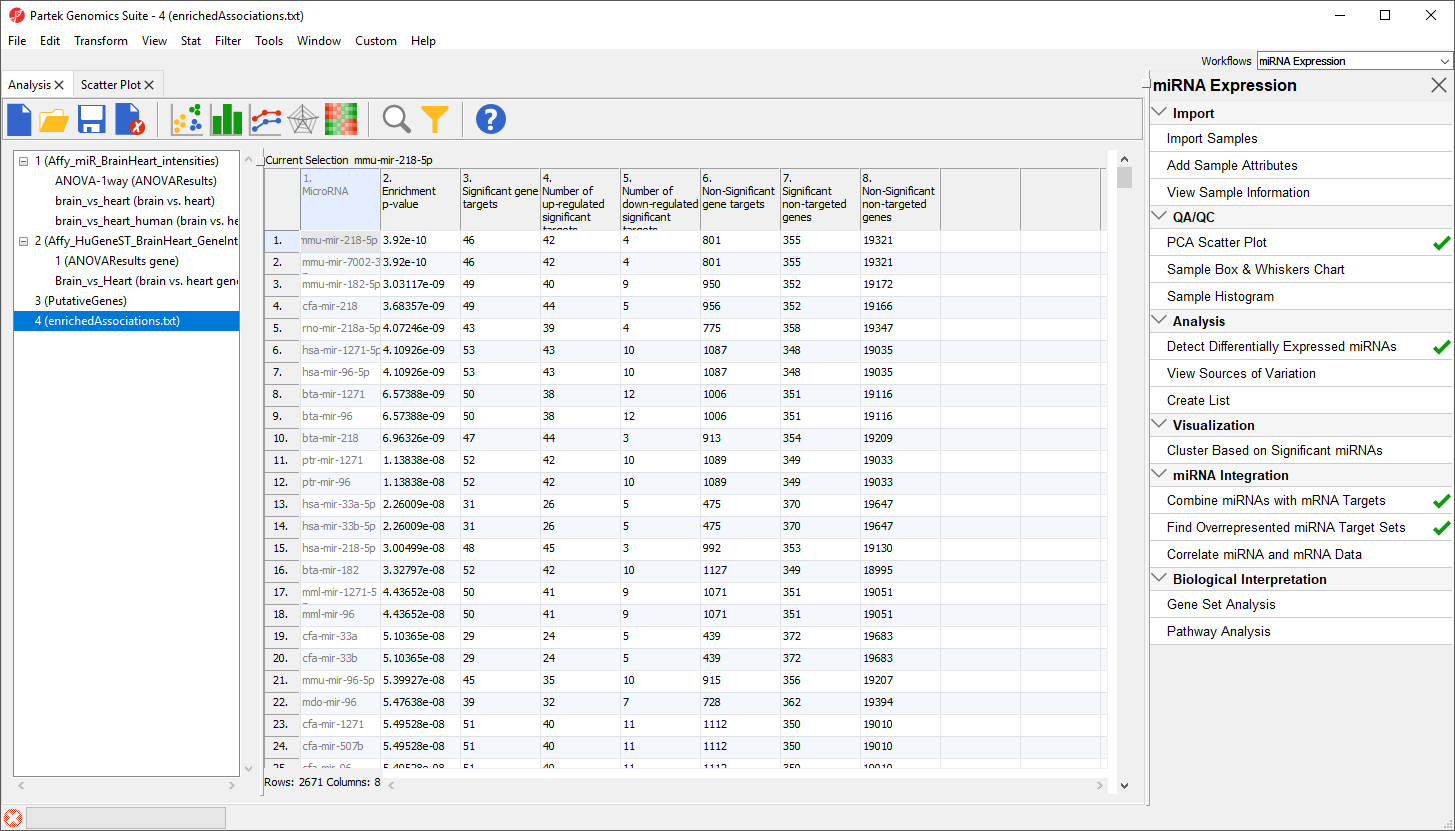
Figure 6. Output of the Find Overrepresented miRNA Target Sets tool
As the enrichment p-values have not been corrected for running multiple statistical tests, we can the multiple test corrrection feature of Partek Genomics Suite to adjust the p-values.
- Select the enrichedAssociations spreadsheet
- Select Stat from the main menu toolbar
- Select Multiple Test Correction
- Select all the multiple test correction options
- Transfer Enrichment p-value to the Selected Column(s) panel from the Candidate Column(s) panel (Figure 7)
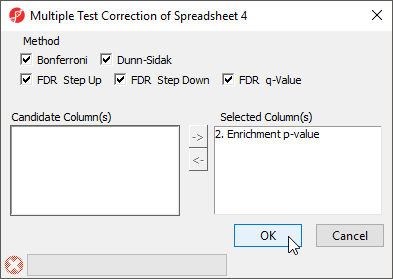
Figure 7. Configuring the Multiple Test Correction dialog
Columns for each of the test correction methods will be added to the enrichedAssociations spreadsheet and can be used to filter the list of miRNAs.
Combine miRNAs with mRNA target genes
This option is useful if you have miRNA and gene expression experiments you want to compare. The samples should be comparable, but do not have to originate from the same specimens.
- Select Combine miRNAs with their mRNA targets from the miRNA Integration section of the workflow
- Select the Get Targets from Spreadsheet tab
- Select TargetScan 7.1 from the Target Database drop-down menu
- Select brain vs. heart human from the microRNA Spreadsheet drop-down menu
- Select 2. Probeset ID for Column with microRNA labels
- Select ANOVAResults gene from the mRNA Spreadsheet drop-down menu
- Select 4. Gene Symbol for Column with gene symbols (Figure 8)
- Select OK
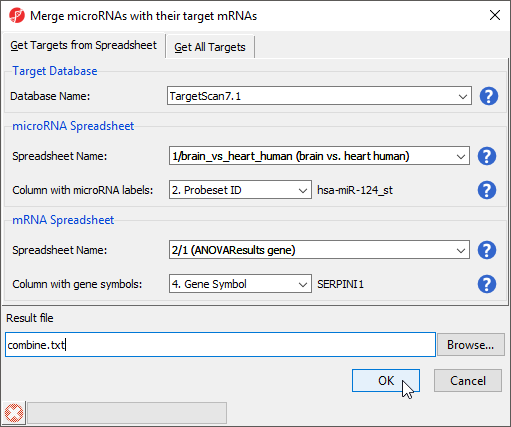
Figure 8. Combining miRNAs with their mRNA targets
In the new spreadsheet, each row represents a specific miRNA associated with one of its target genes; a single miRNA can have multiple targets. For example, hsa-miR-133b_st has 659 rows, one for each target (Figure 9).
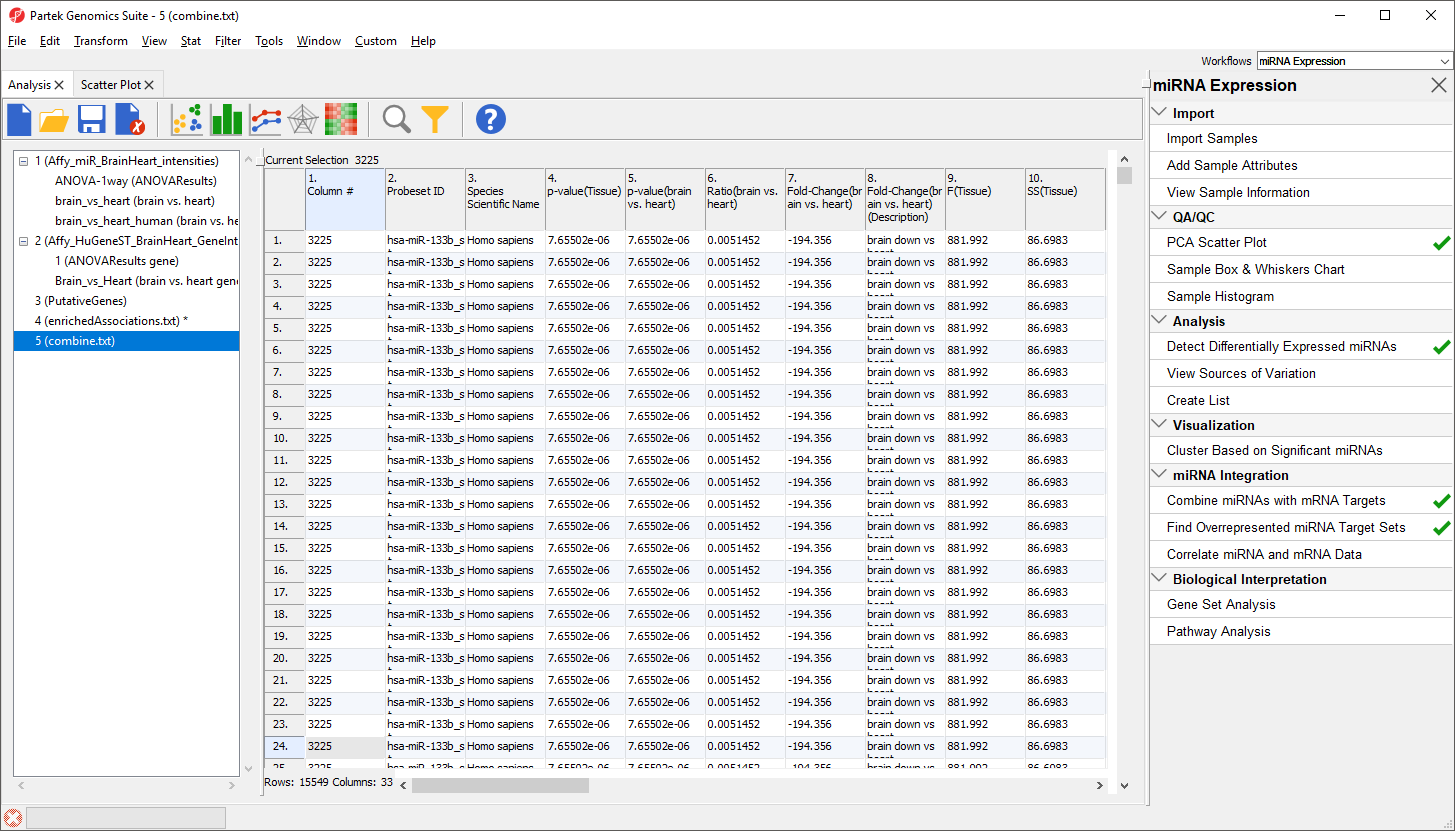
Figure 9. Viewing the combined spreadsheet with miRNAs and mRNA targets
Columns 1-12 are taken from the miRNA expression source spreadsheet while columns 13-26 are taken from the gene expression source spreadsheet.
Correlating miRNA and gene expression data
This application is useful when you have miRNA and mRNA expression data form the same samples and want to correlate the findings to determine whether up- or down-regulated miRNAs result in gene expression changes in their cognate genes. Pearson and Spearman correlation coefficients and their p-values are calculated.
- Select Correlate miRNA and mRNA data from the miRNA Integration section of the workflow
- Select TargetScan7.1 from the Target Database drop-down menu
- Select Affy_miR_BrainHeart_intensities for the microRNA spreadsheet using the drop-down menu
- Select Affy_HuGeneST_BrainHeart_GeneIntensities as the mRNA spreadsheet using the drop-down menu (Figure 10)
- Select OK
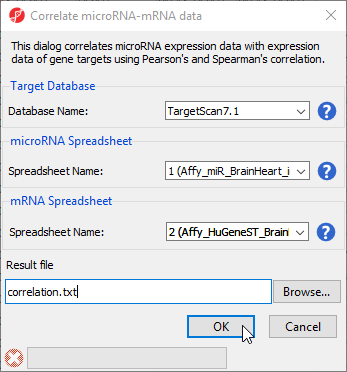 Figure 10. Configuring the Correlate miRNA-mRNA dialog
Next, select the SmapleID column from each spreadsheet. These must match.
Figure 10. Configuring the Correlate miRNA-mRNA dialog
Next, select the SmapleID column from each spreadsheet. These must match.
- Select 6. SampleID for Affy_miR_BrainHeart_intensities
- Select 6. SampleID for Affy_HuGeneST_BrainHeart_GeneIntensities
- Select OK (Figure 11)
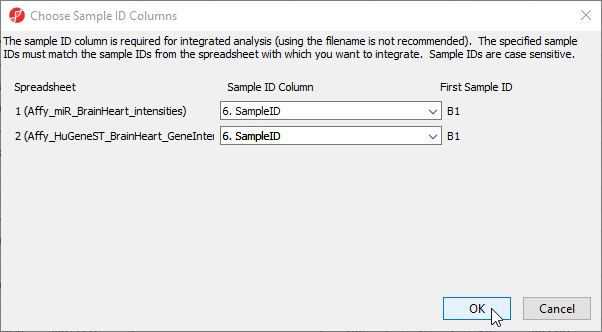
Figure 11. Choosing matching Sample ID columns
The new spreadsheet, correlation.txt (Figure 12). Each row contains one miRNA correlated with one of its target gnees. The first column contains the miRNA probeset ID from the miRNA intensities spreadsheet. The second column contains the mRNA probeset ID from the gene expression intensities spreadsheet. The third column lists the gene symbol and the fourth the miRNA name. The fifth and sixth columns are the Pearson correlation coefficient and its p-value for the gene-miRNA pair. The seventh and eigth columns are the Spearman's rank correlation coefficient and its p-value for the gene-miRNA pair. Negative correlation indicates that a high level of the miRNA is correlated with a low expression level in its target gene. Positive correlation indicates that a high level of the miRNA is associated with a high level of its target gene.
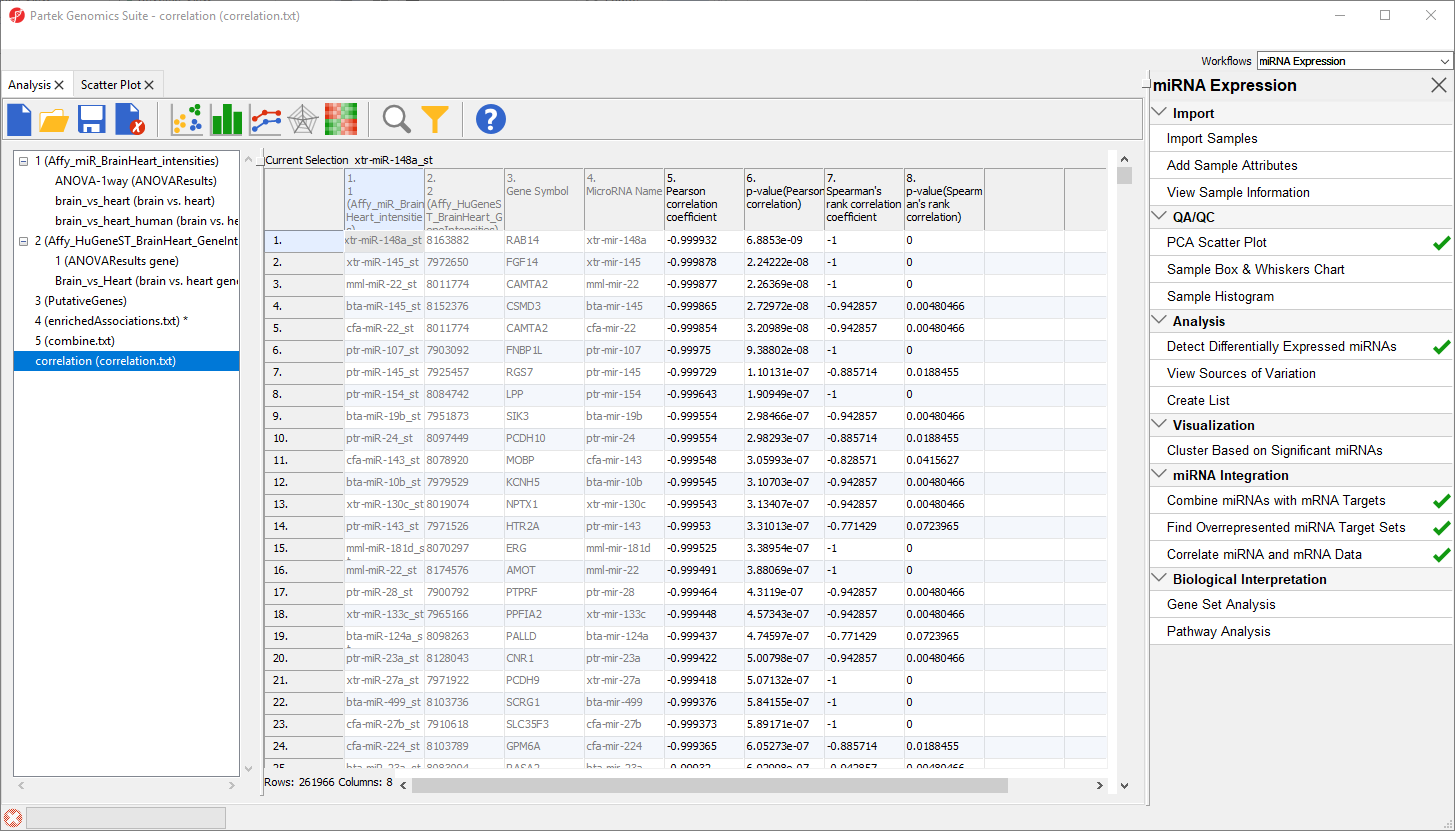
Figure 12. Viewing the correlation spreadsheet
We can visualize the correlation between any miRNA and target gene.
- Right-click a row header
- Select Scatter Plot (Orig. Data) from the pop-up menu
The correlation plot shows miRNA intensitiy on the x-axis and gene expression on the y-axis (Figure 13). Here, we see a negative correlation between expression of xtr-miR-148a_st and its target gene, RAB14, in brain and heart tissues. Drawing the scatter plot will create a temporary file with miRNA and gene expression probe intensities for all samples that is used to draw the plot.
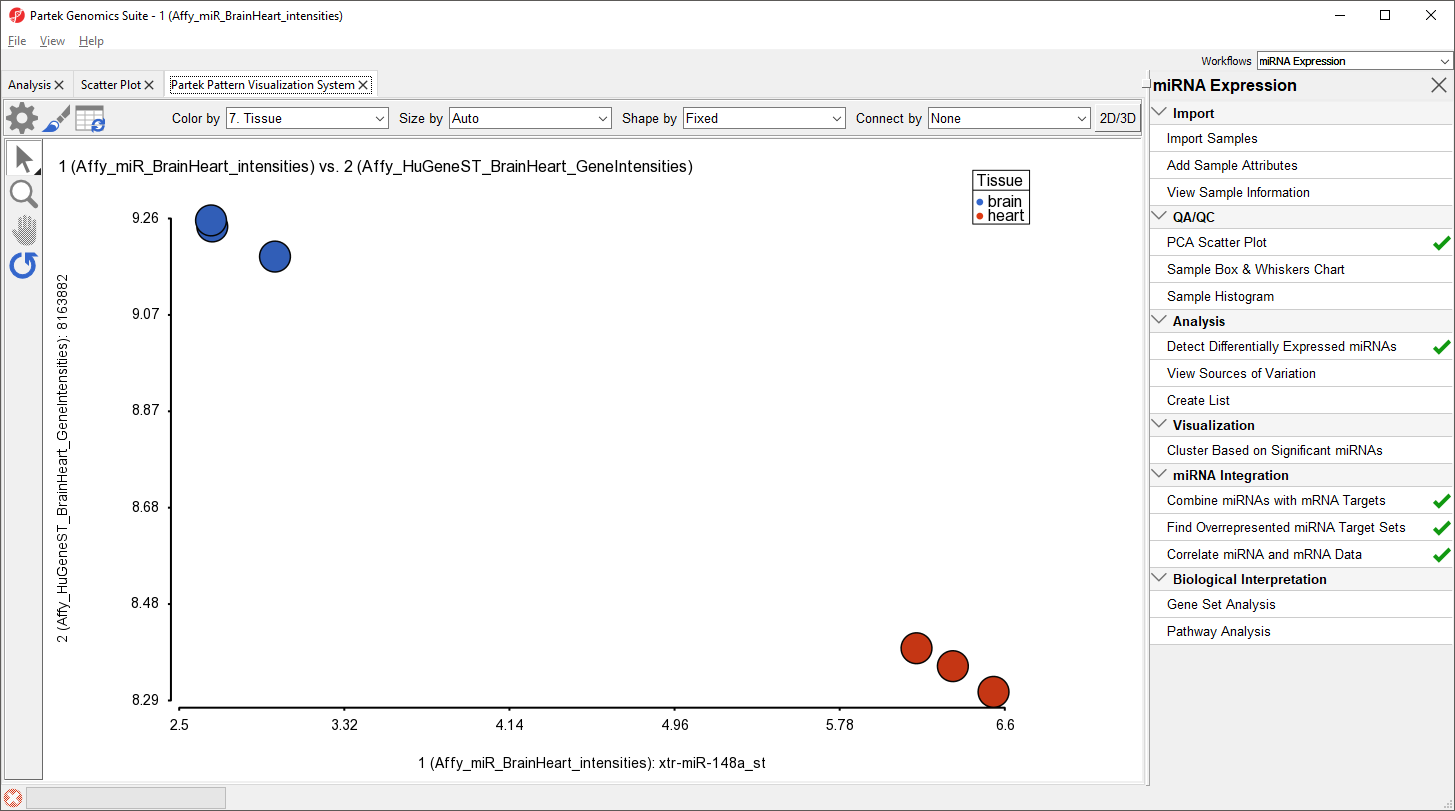
Figure 13. Viewing the scatter plot showing correlated miRNA and target gene expression
Please note that the correlation function is only useful for identifying miRNAs that affect mRNA stability, not translation.
Additional Assistance
If you need additional assistance, please visit our support page to submit a help ticket or find phone numbers for regional support.


| Your Rating: |
|
Results: |

|
33 | rates |
Overview
Content Tools