This option is only available when Cufflinks quantification node is selected. Detailed implementation information can be found in the Cuffdiff manual [5].
When the task is selected, the dialog will display all the categorical attributes more than one subgroups (Figure 1).
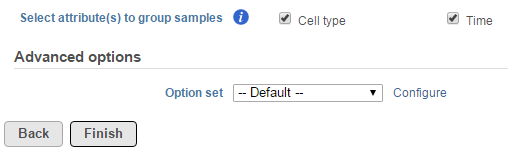
Figure 1. Cuffdiff setup dialog. “Select attributes(s) to groups samples” lists the categorical attributes which have at least two levels (e.g. “Cell type” and “Time”)
When an attribute is selected, pairwise comparisons of all the levels will be performed independently.
Click on Configure button in the Advanced options to configure normalization method and library types (Figure 2).
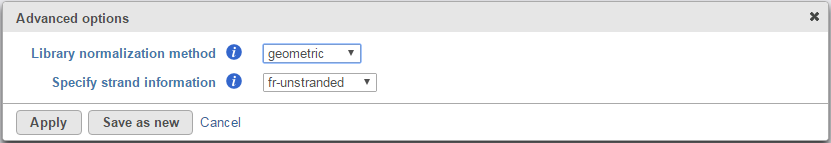
Figure 2. Advanced option of cuffdiff
There are three library normalization methods:
- Class-fpkm: library size factor is set to 1, no scaling applied to FPKM values
Geometric: FPKM are scaled via the median of the geometric means of the fragment counts across all libraries [6]. This is the default option (and is identical to the one used by DESeq)
- Quartile: FPKMs are scaled via the ratio of the 75 quartile fragment counts to the average 75 quartile value across all libraries
The library types have three options:
- Fr-unstranded: reads from the left-most end of the fragment in transcript coordinates map to the transcript strand, and the right-most end maps to the opposite strand. E.g. standard Illlumina
- Fr-firststrand: reads from the left-most end of the fragment in transcript coordinates map to the transcript strand, and the right-most end maps to the opposite strand. The right-most end of the fragment is the first sequenced or only sequenced for single-end reads. It is assumed that only the strand generated during first strand synthesis is sequenced. E.g. dUPT, NSR, NNSR
- Fr-secondstrand: reads from the left-most end of the fragment in transcript coordinates map to the transcript strand, and the right-most end maps to the opposite strand. The left-most end of the fragment is the first sequenced or only sequenced for single-end reads. It is assumed that only the strand generated during second strand synthesis is sequenced. E.g. Directional Illumina, standard SOLiD.
The report of the cuffdiff task is a table of a feature list p-values, q-value and log2 fold-change information for all the comparisons (Figure 20).
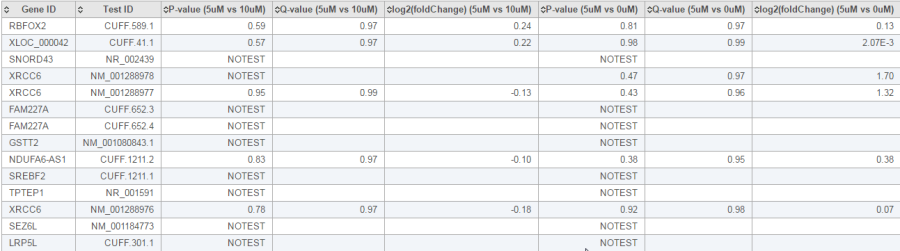
Figure 3. Figure 20: Cuffdiff task report. Each row is a feature, p-value, q-value and log2 fold change columns are display for each comparison
In the p-value column, besides an actual p-value, which means the test was performed successfully, there is also the following flags which indicate the test was not successful:
- NOTEST: not enough alignments for testing
- LOWDATA: too complex or shallowly sequences
- HIGHDATA: too many fragments in locus
- FAIL: when an ill-conditioned covariance matrix or other numerical exception prevents testing
The table can be downloaded as a text file when clicking the Download button on the lower-right corner of the table.
References
- Benjamini, Y., Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing, JRSS, B, 57, 289-300.
- Storey JD. (2003) The positive false discovery rate: A Bayesian interpretation and the q-value. Annals of Statistics, 31: 2013-2035.
- Auer, 2011, A two-stage Poisson model for testing RNA-Seq
- Burnham, Anderson, 2010, Model selection and multimodel inference
- Law C, Voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biology, 2014 15:R29.
- http://cole-trapnell-lab.github.io/cufflinks/cuffdiff/index.html#cuffdiff-output-files
- Anders S, Huber W: Differential expression analysis for sequence count data. Genome Biology, 2010
Additional Assistance
If you need additional assistance, please visit our support page to submit a help ticket or find phone numbers for regional support.


| Your Rating: |
|
Results: |

|
40 | rates |
Overview
Content Tools
1 Comment
Melissa del Rosario
author: wxw