Salmon1 is a method for quantifying transcript abundance from raw read sequence fastq files, it will generate transcript level count matrix as output.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5600148/
Running Salmon
Salmon will run on any FASTQ file input.
- Click the data node containing your FASTQ files
- Click the Quantification section in the toolbox
- Click Salmon
- Choose Assembly and Aligner index on gene annotation model (Figure 1)
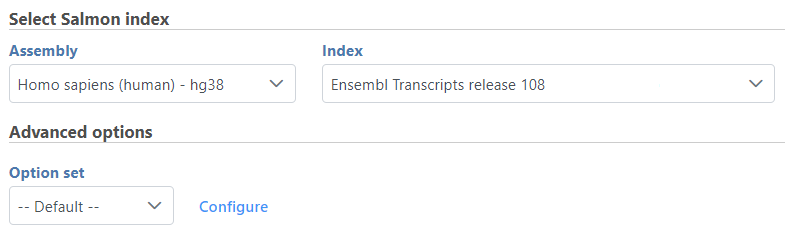
Figure 1. Configure Salmon task--select index file
Salmon index can't be built on reference assembly, you need to provide transcript annotation file beforehand, the index will be built on transcript annotation model. For more information about adding aligner index based on annotation model, see this chapter in library file management.
The task generates two data nodes: Transcript counts node contains NumReads which is the estimate of number of reads mapping to each transcript; the best estimate is often not integer. Gene counts node contains the sum of all the reads from the corresponding transcripts from each gene.
Note: If you want to perform differential analysis, you need to add offset to deal with 0 values.
References
Rob Patro, Geet Duggal, Michael Love, Rafeal A Irizarry, Carl Kingsford. Salmon: fast and bias-aware quantification of transcript expression using dual-phase inference. Published online 2017 Mar 6. doi:10.1038/nmeth.4197
Additional Assistance
If you need additional assistance, please visit our support page to submit a help ticket or find phone numbers for regional support.


| Your Rating: |
|
Results: |

|
11 | rates |
Overview
Content Tools