With the Partek Flow REST API, you can create custom solutions to query or drive your server. Below are some common use cases for the REST API:
A complete reference for the API can be found on the REST API Command List or by visiting [server]/api/v1/servlets
The referenced Python library can be downloaded here.
Generate an authentication token
An access token can be generated from the System information section of the settings page.

Alternatively, GetToken.py will generate a token:
python GetToken.py --server localhost:8080 --user admin
you will be prompted to enter your password.
This token can be specified as the token parameter.
curl --form token=cUOWY0VvkSFagr... http://localhost:8080/flow/api/v1/users/list
Create a project
Flow organizes data by projects and they can be created and managed by the REST API.
To create a project:
curl -X POST --form token=$FLOW_TOKEN --form project="My Project" http://localhost:8080/flow/api/v1/projects
The server will respond with JSON data describing the new project:
{"name":"My Project","id":"0","description":"","owner":"0","userRoles":{"0":"Project owner"},"outputFolders":{"0":"/home/flow/FlowData/Project_My Project"},"diskUsage":"0 GB","lastModifiedTimeStamp":1506013662476,"lastModifiedDate":"12:00 PM","data":[]}
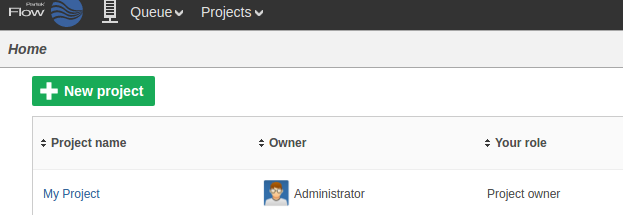
The new project will appear on the Flow homepage:
Upload a group of samples
UploadSamples.py is a python script that can create samples within a project by uploading files:
python UploadSamples.py --verbose --token $FLOW_TOKEN --server http://localhost:8080 --project "My Project" \ --files ~/MoreData/REST/sample1.fastq.gz ~/MoreData/REST/sample2.fastq.gz ~/MoreData/REST/sample3.fastq.gz ~/MoreData/REST/sample4.fastq.gz
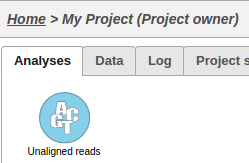
This operation will generate a data node on the Analyses tab for the imported samples:
Assign sample attributes
We can associate attributes with samples for use in visualizations and statistical analysis:
python AddAttribute.py -v --server http://localhost:8080 --token $FLOW_TOKEN --project_name "My Project" --sample_name sample1 --attribute Type --value Case python AddAttribute.py -v --server http://localhost:8080 --token $FLOW_TOKEN --project_name "My Project" --sample_name sample2 --attribute Type --value Case python AddAttribute.py -v --server http://localhost:8080 --token $FLOW_TOKEN --project_name "My Project" --sample_name sample3 --attribute Type --value Control python AddAttribute.py -v --server http://localhost:8080 --token $FLOW_TOKEN --project_name "My Project" --sample_name sample4 --attribute Type --value Control
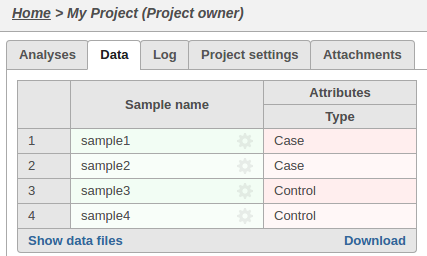
The sample attributes can be viewed and managed on the data tab:
Run a pipeline
A pipeline is a series of tasks used to process and analyze genomic data. You can read more about pipelines here
To run a pipeline, first we need to know its name.
We can get the name of a pipeline from the GUI or from the API:
wget -q -O - http://localhost:8080/flow/api/v1/pipelines/list$AUTHDETAILS | python -m json.tool | gvim -
Many pipelines also require that library files are specified.
You can get the list of required inputs for the pipeline from the API:
http://localhost:8080/flow/api/v1/pipelines/inputs?project_id=0&pipeline=AlignAndQuantify
This particular pipeline requires a bowtie index and an annotation model:

The request to launch the pipeline needs to specify one resource ID for each input.
These IDs can be found using the API:
Get the IDs for the library files that match the required inputs
wget -q -O - "http://localhost:8080/flow/api/v1/library_files/list${AUTHDETAILS}&assembly=hg19" | python -m json.tool | gvim -
[
{
"annotationModel": "",
"assembly": "hg19",
"description": "Reference sequence",
"fileType": "Genome sequence",
"id": 100
},
{
"annotationModel": "",
"assembly": "hg19",
"description": "Cytoband",
"fileType": "cytoBand.txt",
"id": 101
},
{
"annotationModel": "",
"assembly": "hg19",
"description": "Bowtie index",
"fileType": "Bowtie Index",
"id": 102
},
{
"annotationModel": "hg19_refseq_15_05_07_v2",
"assembly": "hg19",
"description": "Annotation file: hg19_refseq_15_05_07_v2",
"fileType": "Annotation model",
"id": 103
}
]
The pipeline can be launched in any project using RunPython.py
python RunPipeline.py -v --server http://localhost:8080 --token $FLOW_TOKEN --project_id 0 --pipeline AlignAndQuantify --inputs 102,103
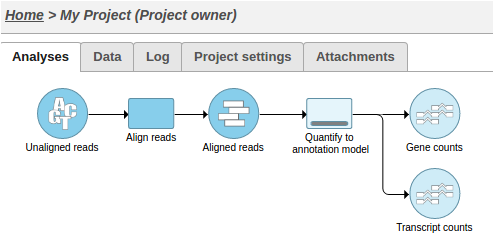
This action will cause two tasks to start running:
Alternatively, UploadSamples.py can create the project, upload the samples and launch the pipeline in one step:
python UploadSamples.py -v --server http://localhost:8080 --token $FLOW_TOKEN --files ~/sampleA.fastq.gz ~/sampleB.fastq.gz --project NewProject --pipeline AlignAndQuantify --inputs 102,103
Add a collaborator to a project
To add a collaborator to a project:
curl -X PUT "http://localhost:8080/flow/api/v1/projects?project=ProjectName&collaborator=user1&role=Collaborator&token=$FLOW_TOKEN"
Transfer feature lists
curl --form token=$TO_TOKEN --form url=http://from:8080/flow/api/v1/feature_lists/export?token=$FROM_TOKEN http://to:8080/flow/api/v1/feature_lists/import
Monitor a folder and upload files as they are created
#!/bin/bash
inotifywait -m $PATH_TO_MONITOR -e create -e moved_to |
while read path action file; do
if [[ $file == *.fastq.gz ]]; then
echo "Uploading $file"
python UploadSamples.py -v --server $SERVER --token $FLOW_TOKEN --files $path/$file --project "$PROJECT"
fi
done
Monitor the queue and send a notification if there are too many waiting tasks
#!/bin/bash
while true; do
result=`python QueueStatistics.py --server $SERVER --token $TOKEN --max_waiting $MAX_WAITING`
if [ $? -eq 1 ]; then
/usr/bin/notify-send $result
exit 1
fi
sleep $INTERVAL
done
Additional Assistance
If you need additional assistance, please visit our support page to submit a help ticket or find phone numbers for regional support.


| Your Rating: |
|
Results: |

|
13 | rates |
Overview
Content Tools