Cell Hashing enables sample multiplexing and super-loading in single cell RNA-Seq isolation by labeling each sample with a sample-specific oligo-tagged antibody against a ubiquitously expressed cell surface protein.
The Hashtag demultiplexing task is an implementation of the algorithm used in Stoeckius et al. 20181 for multiplexing cell hashing data. The task adds cell-level attributes Sample of origin and Cells in droplet.
Prerequisites for running Hashtag demultiplexing
To run Hashtag demultiplexing, your data must meet the following criteria:
- Data node contains number of features less than number of observations
- Data node must be output from normalization task (recommended normalization method for hashtag is CLR)
If you are processing your FASTQ files in Partek Flow, be sure to specify a different Data type for your Cell Hashing FASTQ files on import than the FASTQ files for your gene expression and any other antibody data.
If you are processing your FASTQ files using Cell Ranger, be sure to specify a different feature_type for your Cell Hashing antibodies than any other antibodies in the Feature Reference CSV File.
If you want to specify sample IDs instead of using hashtag feature IDs as the sample IDs, you will need to prepare a tab-delimited text file (.txt) with hashtag feature IDs in the first column and the corresponding sample IDs in the second column (Figure 1). A header row is required.
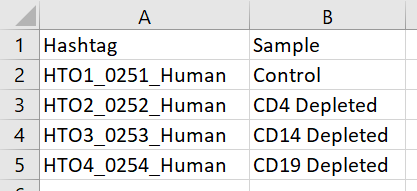
Figure 1. Sample ID .txt file example. The first column is the hashtag feature ID and the second column is the corresponding sample ID
Running Hashtag demultiplexing
- Click the Normalized counts data node for your cell hashing data
- Click Hashtag demultiplexing in the Pre-analysis tools section of the toolbox
- Click Browse to select your Sample ID file (Optional)
- Click Finish to run
The output is a Dumultiplexed counts data node (Figure 2).
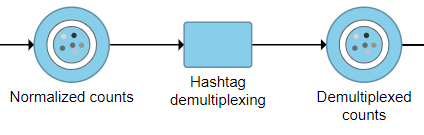
Figure 2. Hashtag demultiplexing can be run on a Normalized counts data node and outputs a Demultiplexed counts data node
Two cell-level attributes, Cells in droplet and Sample of origin, are added by this task and are available for use in downstream tasks.You can download the attribute values for each cell by clicking the Demultiplexed counts data node, clicking Download, and choosing to download Attributes only.
We recommend using Annotate cells to transfer the new attributes to other sections of your project after downloading the attributes text file.
It is also possible to use the Merge matrices task to combine your data types and attributes.
References
- Stoeckius, M., Zheng, S., Houck-Loomis, B., Hao, S., Yeung, B.Z., Mauck, W.M., Smibert, P. and Satija, R., 2018. Cell Hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome biology, 19(1), p.224.
Additional Assistance
If you need additional assistance, please visit our support page to submit a help ticket or find phone numbers for regional support.


| Your Rating: |
|
Results: |

|
26 | rates |
Overview
Content Tools