Controls
Chromosome view can be customized by using the control panel on the left (Figure 1). The Attribute and Order By controls show options depending on the current project, while the content of the Annotate amino acids control depends on the annotation files associated with the current genome build in the Library File Management. In order for any change to take place, push the Apply button.
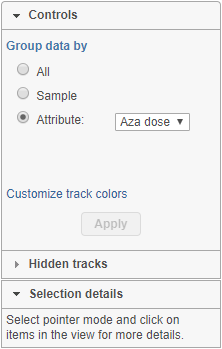
Figure 1. Control panel (an example is shown)
Group data by
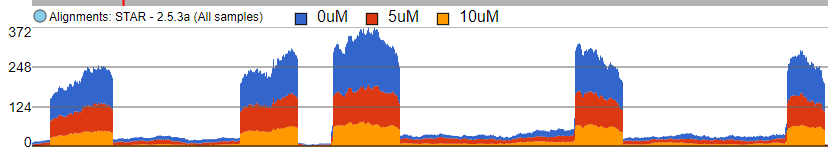
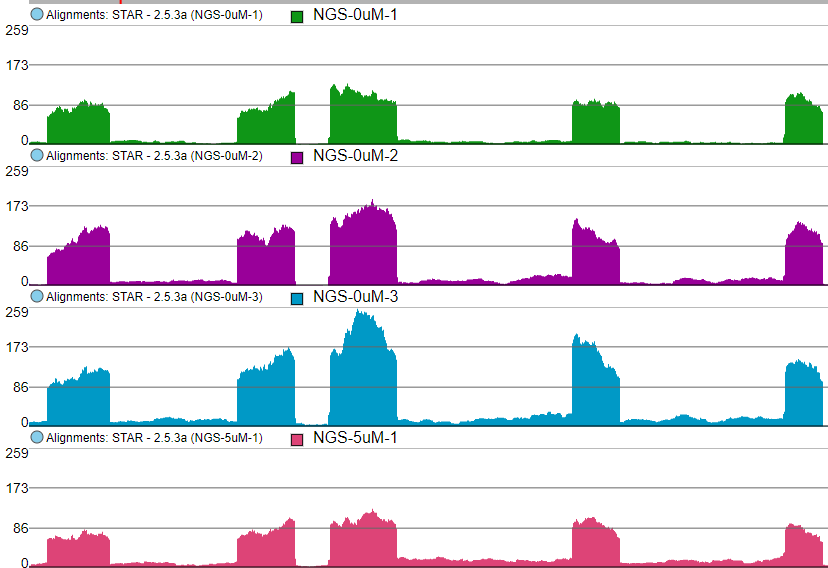
The first option, Group data by, specifies the number of Alignments tracks (Figure 2). All will result in only one track, with all the samples on it. Sample creates one track per sample, while Attribute produces one Alignments track per level of the Attribute (i.e. one track per group).
All
Sample
Attribute
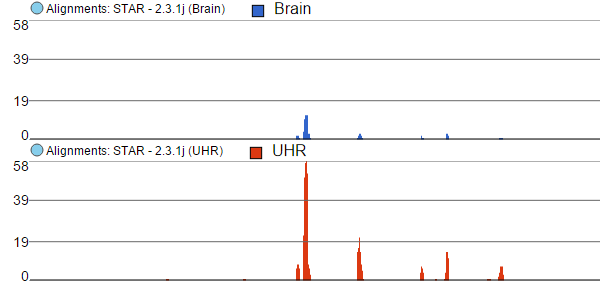
Figure 2. Group data by: All creates one Alignments track for the entire project, Sample creates one Alignments track for each sample, Attribute creates one Alignments track for each group (an example is shown)
Annotate amino acids by
Annotate amino acids by controls the appearance of the Amino acids track and allows you to pick the transcript database that will be used to plot codons (Figure 3). The drop down list shows the databases currently available for the selected genome (additional databases can be added via Library File Management).
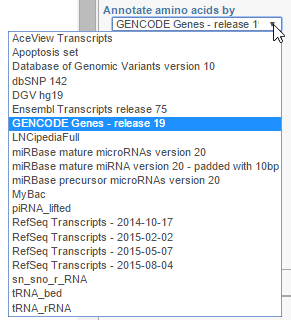
Figure 3. Annotate amino acids by: transcript models currently associated with the chosen genome are displayed in the drop-down list and can be used to plot Amino acids track (an example is shown)
Color by
Color by option affects the colouring of the Alignments track and Isoform proportion track. When Sample is selected from the drop-down list, individual samples will be shown on the aforementioned tracks, each sample being given a different colour. If attributes were assigned to samples, they will also be visible in the Color by drop-down (Figure 4) and you will be able to highlight levels of the selected attribute (Figure 5).
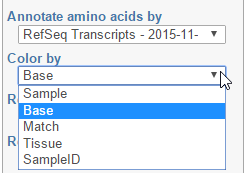
Figure 4. Color by: the options control colouring of Alignments and Isoform proportion tracks. Sample, Base, and Match options are present by default. If attributes have been assigned to samples, they will appear in the drop-down list. In this example, that is the "Tissue" attribute
Color by Sample
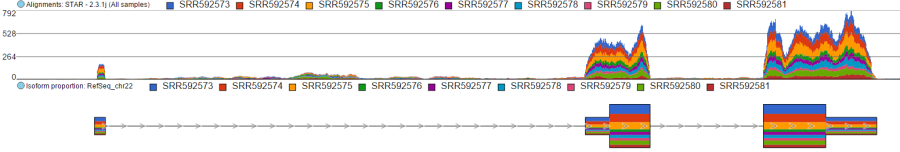
Color by <Attribute>
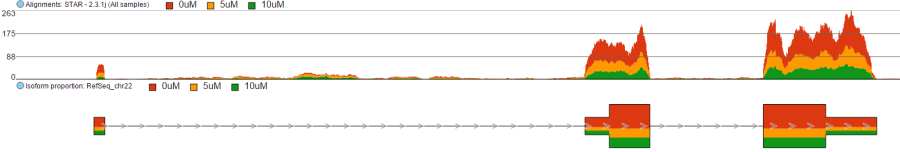
Figure 5. Difference between Color by Sample and Color by . Color by Sample uses different colours to depict individual samples; Color by uses different colours to depict levels of the selected sample attribute (as present in the Data tab). Alignments and Isoform proportion tracks are shown (an example)
The effect of the option to Color by Base can be seen with high power magnification (Figure 6). Individual base calls are highlighted by different colours. When that option is chosen at low power magnification, all the bases are shown in grey.

Figure 6. Color by Base highlights the base calls by colours. Different colours are visible with high power magnification; otherwise all the bases are shown in gray (an example)
Finally, Color by Match can be used to quickly identify mismatches against the reference genome. A matching base is coloured in blue, while mismatch bases are shown in yellow.
Read histogram Y-axis max
The maximum of the y-axis of Alignments tracks is set by Read histogram Y axis scales option (Figure 7). When using Project max, the y-axis for each track is set individually, based on the maximum within that sample. On the other hand, Track max uses the maximum across all the samples and uses that value as the maximum for all.
Project max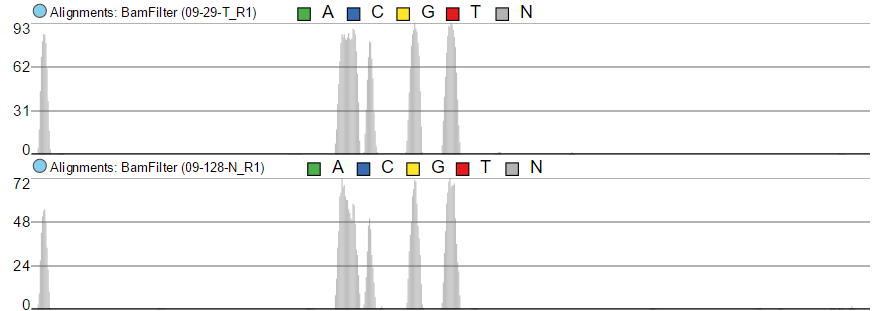
Track max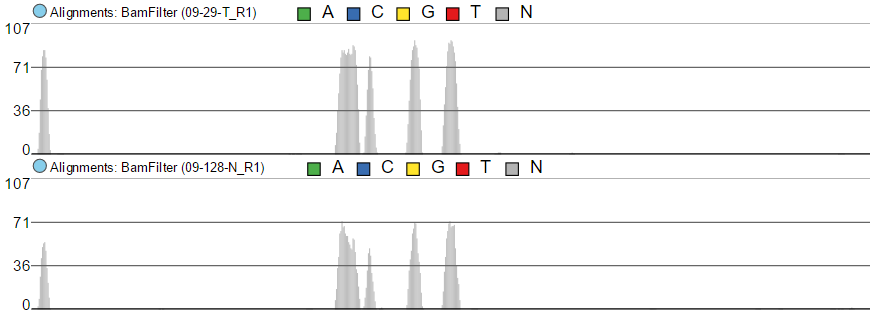
Figure 7. Read histogram Y axis scales. When set to Linked, all the tracks have the same Y axis maximum, which depends on the sample with the highest coverage. Using Independent sets Y axis maximum independently for each sample.
Read histogram type
Read histogram type changes the presentation of the Alignments track and should be used in conjunction with the Group data by and Color by tracks to get the desired visualisation.
When set to Sum, the Read histogram type shows the sum of base calls at each position, i.e. total coverage per position. Figure 8 shows an Alignments track with three samples. With the Sum option, the number of reads at each base in each sample is added and displayed. The contribution of individual samples is not visible since the track is Colored by Group (but that would make sense in this example).

Figure 8. Alignments track: total coverage per locus is shown by using "Read histogram type" set to "Sum" and "Group data by" set to
To show the average coverage per locus, switch Read histogram type to Average and leave Color by as is (i.e. by group) (Figure 9). With this setting, Chromosome view will calculate the average by dividing the total coverage per locus by the number of samples. Note that using Color by Sample would not make sense here. Although Figure 8 looks quite like Figure 7, the y-axis range is different.

Figure 9. Alignments track: average coverage per locus is shown by using "Read histogram type" set to "Average", "Group data by" set to "Attribute", and "Color by" set to
Finally, the option Overlay is useful if you want to directly compare base counts over several samples (or groups) as each will be represented by a line (i.e. no stacking). The example in Figure 10 is based on microarray data, showing three groups on the same Alignments track. The red group has the highest base counts, while the counts in the blue group are much lower.
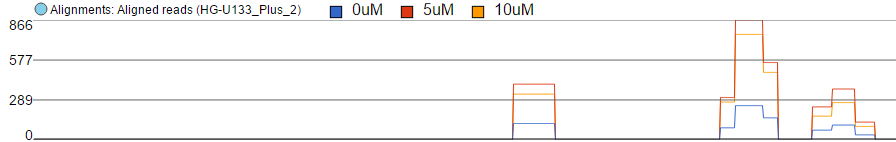 Figure 10. Alignments track: coverage per locus is shown by using "Read histogram type" set to "Overlay". Each plot is a single experimental condition ("Group data by" set to "Attribute", "Color by" set to
Figure 10. Alignments track: coverage per locus is shown by using "Read histogram type" set to "Overlay". Each plot is a single experimental condition ("Group data by" set to "Attribute", "Color by" set to
Split read histogram by strand
To view the reads histogram grouped by the specific strand that they've mapped to, click the Split read histogram by strand checkbox. This displays the forward reads at the top half of the track and the reverse reads on the bottom half of the track (Figure 11). This track can be helpful in studies such as ChIP-Seq, where strand-specific read distributions can display hallmarks of DNA-protein interactions.
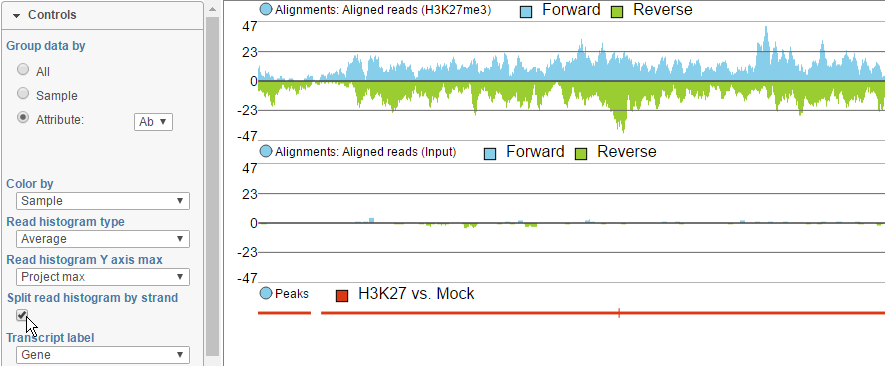 Figure 11. Viewing reads by strand along the reads histogram
Figure 11. Viewing reads by strand along the reads histogram
Transcript label
You can use the Transcript label selector to specify labels on the reference transcript track and Isoform proportion track (Figure 12).
Transcript label: Gene![]()
Transcript label: Transcript![]()
Figure 12. ranscript label: setting the control to Gene shows only gene label, while Transcript shows transcript labels. Both transcript database and Isoform proportion tracks are affected Short sequencing reads can be coloured by strand (Reads pileup color: Strand) or by base (Reads pileup color: Base).
Reads pileup and probe color
Reads pileup color: Strand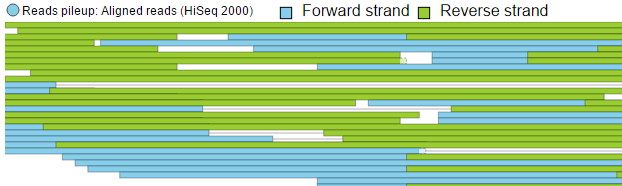
Reads pileup color: Base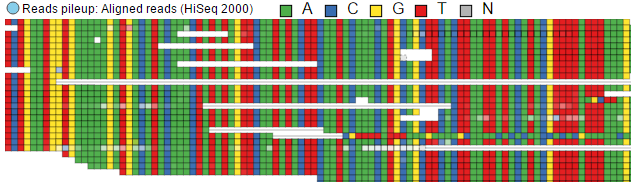
Figure 13. Reads pileup color: colouring of the short sequencing reads by Strand or by Base
Probe color control customizes the appearance of Probe intensities track (Figure 14). When set to Intensity, the color of a probe reflects its intensity, using a color gradient from white (low) to admiral (high). Alternatively, when Strand is turned on, probes on the reverse strand are in parakeet green, while probe on the forward strand is in sky blue.
Probe color: Intensity
Probe color: Strand
Figure 14. Probe color: "intensities" colors probes proportionally to their intensity, "strand" uses colors to indicate probe positioning (an example is shown)
If a variant database is available for the current genome, the variants can be added to the Reference genome track. To show the variants, point the Variant database control to the database of your choice.
To change any of the colors on the canvas, use the Customize track colors tool. A resulting dialog will help you to pick another color (drop-down button opens the color-picker) (Figure 15).
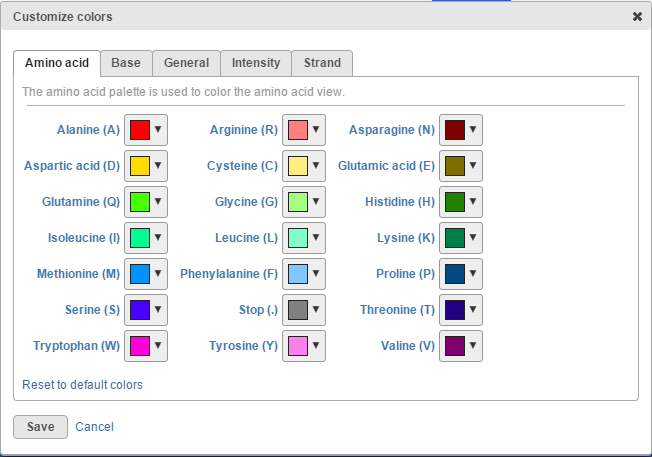
Figure 15. Customize colors dialog: selecting a drop-down arrow opens the color-picker tool
Track Order
The position of the tracks on canvas can be controlled by using the Track order tool. If you want a track to be visible all the time, i.e. while scrolling up or down, pin it to the top or to the bottom. Below shows the Cytoband track pinnned to the top of the canvas and Reference genome track pinned to the bottom of the canvas. To unpin a track, click on the pin icon ( ![]() ). The track will be unpinned and a message No tracks are pinnned to the top / bottom will appear. To pin a track, drag the track name to the No tracks… message. Alternatively, you can use the green arrows (
). The track will be unpinned and a message No tracks are pinnned to the top / bottom will appear. To pin a track, drag the track name to the No tracks… message. Alternatively, you can use the green arrows ( ![]() ) to pin a track. When you mouse over an arrow, the new position of the track will be highlighted on the canvas; click on the arrow to accept.
) to pin a track. When you mouse over an arrow, the new position of the track will be highlighted on the canvas; click on the arrow to accept.
A track can be hidden (meaning it will not be visible) by selecting the red minus, or unhidden by selecting the green plus icon.
The tracks can be reordered by drag and drop.
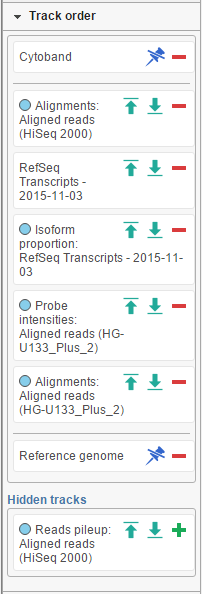
Figure 16. Track order tool: To change the position of a track drag and drop to the new position. To pin a track to the top / bottom of the canvas, use the up and down arrows. To unpin a track, select the pin icon. A track can be hidden by clicking on the red minus symbol and unhidden by selecting the green plus. Coloured dot by a track names indicates the layers to which the track belongs (an example is shown)
Selection Details
At the bottom of the control panel you will find the Selection details section (Figure 17). It is used to display information on the element selected on the canvas (using the Pointer mode).
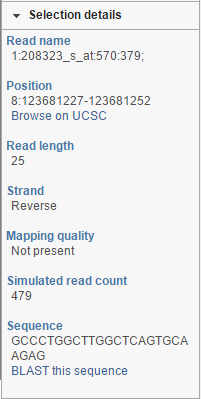
Figure 17. Selection details showing information on the element selected on the canvas. The example shows details of a microarray probe. Note the two link-outs ("Browse on UCSC" and "BLAST this sequence")
Additional Assistance
If you need additional assistance, please visit our support page to submit a help ticket or find phone numbers for regional support.


| Your Rating: |
|
Results: |

|
35 | rates |
Overview
Content Tools
1 Comment
Melissa del Rosario
author: ilukic