Aligned reads can be converted to unaligned reads in Partek Flow. The task is available under Post-alignment tools in the task menu when any Aligned reads data node is selected, which can be a result of an aligner in Partek Flow or data already aligned before import.
Generating unaligned reads from aligned data gives you the flexibility to remap the reads using either a different aligner, a different set of alignment parameters, or a different genome reference. This is particularly useful in analyzing sequences from xenograft models where the same set of reads can be aligned two different species. It may also be useful if the original unaligned FASTQ files are not as easily accessible to the user as the aligned BAM files.
To perform the task, select an Aligned reads data node and click Convert alignments to unaligned reads task in the task menu (Figure 1).
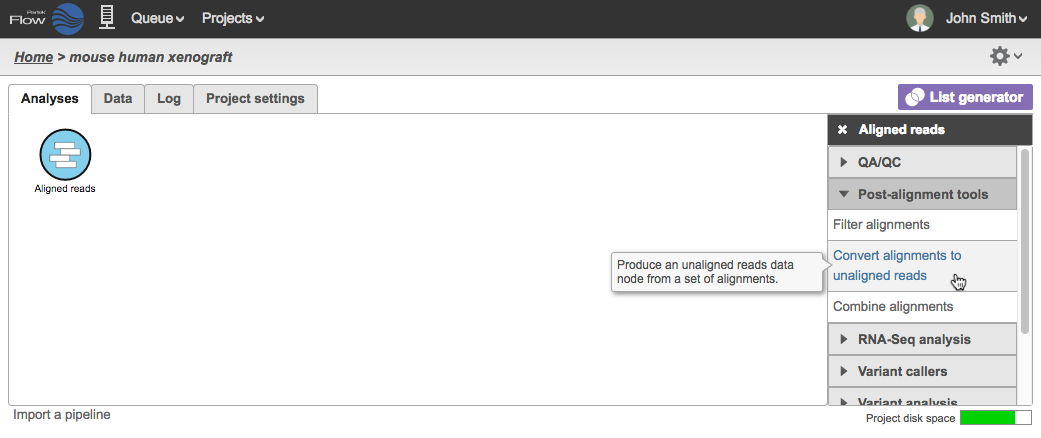
Figure 1. Convert alignments to unaligned reads
During the conversion, the BAM files are converted to FASTQ files and a new Unaligned reads data node will be generated (Figure 2) .
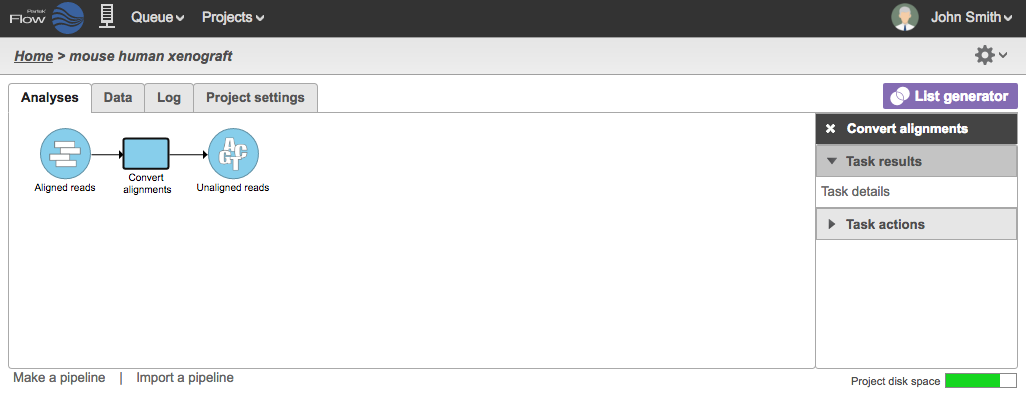
Figure 2. Unaligned reads generated from aligned reads
The filenames of the FASTQ files will be based on the sample names in the Data tab. The files generated are compressed with the extension *.fq.gz. For samples containing BAM files with paired end reads, two FASTQ files will be generated for each, and the files names will be appended with _1 and _2. An example in Figure 3 shows 18 .fq.gz output files that came from 9 BAM files.
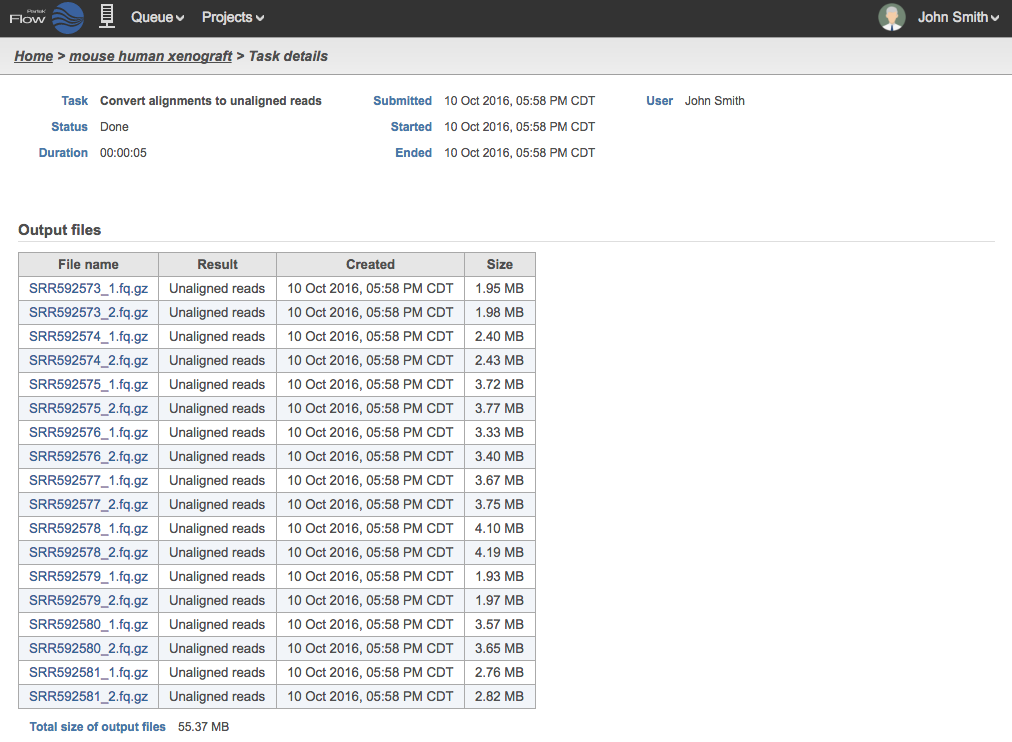
Figure 3. Task details of unaligned reads generated from aligned paired end samples
Additional Assistance
If you need additional assistance, please visit our support page to submit a help ticket or find phone numbers for regional support.


| Your Rating: |
|
Results: |

|
43 | rates |
Overview
Content Tools
1 Comment
Melissa del Rosario
author: mdelrosario