Cufflinks assembles transcripts and estimates transcript abundances on aligned reads. Implementation details are explained in Trapnell et al. [1]
The Cufflinks task has three options that can be configured (Figure 1):
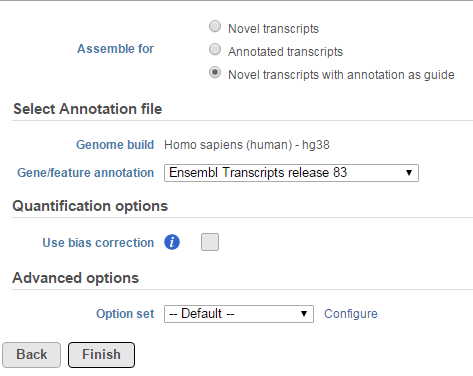
Figure 1. Cufflinks configuration dialog
- Novel transcript: this option does not require any annotation reference, it will do de novo assembly to reconstruct transcripts and estimate their abundance
- Annotation transcript: this option requires an annotation model to quantify the aligned reads to known transcripts based on the annotation file.
- Novel transcript with annotation as guide: this option requires an annotation file to quantify the aligned reads to known transcripts as well as assemble aligned reads to novel transcripts. The results include all transcripts in the annotation file plus any novel transcripts that are assembled.
When the Use bias correction check box is selected, it will use the genome sequence information to look for overrepresented sequences and improve the accuracy of transcript abundance estimates.
References
- Trapnell C, Williams B, Pertea G, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature Biotech. 2010; 28:511-515.
Additional Assistance
If you need additional assistance, please visit our support page to submit a help ticket or find phone numbers for regional support.


| Your Rating: |
|
Results: |

|
43 | rates |
Overview
Content Tools