Peak calling task is used to detect enriched genomic regions on reads generated from ChIP-seq, DNase-seq, MeDIP-seq etc. experiments. Partek® Flow® provides a widely used method MACS2--model-based analysis1 (http://liulab.dfci.harvard.edu/MACS/) to find peaks , it can be used with or without control sample.
MACS2 dialog
Selecting MACS2 from the context sensitive menu will bring up the MACS2 task dialog, interface will be different depends the input aligned data node.
whether there are sample attributes available in the data tab.
If the selected aligned data node was imported, reference assembly the data aligned to needs to be specified, choose the assembly the sequence aligned to from the drop-down list (Figure 1); if the alignment was performed in Partek Flow, this option will not appear.
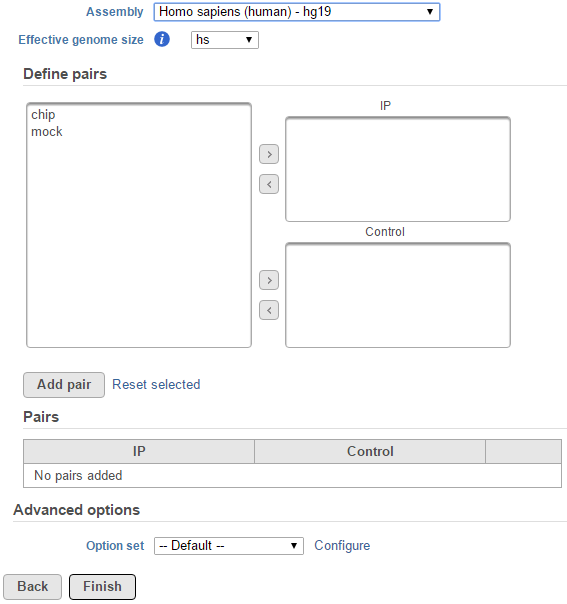 Figure 1. MACS2 dialog: manually add ChIP vs control pairs for peak detection
Figure 1. MACS2 dialog: manually add ChIP vs control pairs for peak detection
The effective genome size is the genome size can be sequenced. Because of the repetitive features on the chromosomes, the actual mappable genome size will be smaller than the original size, typically about 70%-90% of the genome size. There are presets of 4 species based on MACS2 recommendation1 for this parameter:
hs – human, size is 2.7e9
mm – mouse, size is 1.87e9
ce – C. elegans, size is 9e7
dm – fruit fly, size is 1.2e8
When Other is selected, a specific value of the effective genome size needs to be specified in bps as unit (Figure 2).
![]() Figure 2. Specify other species effective genome size by manually type in the value
Figure 2. Specify other species effective genome size by manually type in the value
When sample attribute is not specified, for instance there are only two sample -- ChIP and mock as sample name, the peak detection pairs needs to be manually defined (Figure 1).
In the Define pairs section, the left panel list all the sample names, add one pair at a time, select ChIP sample to put in IP panel on the top-right, choose control sample to put in the Control panel on the bottom-right (Figure 1). If there is no control sample in the experiment, the Control panel can be blank. If more than one ChIP or Control samples added, the samples will be combined (or pooled) in the analysis.
If the sample attributes are defined, you will have an additional option to add pairs based on the attribute. For instance Figure 3 is show an example data with 4 samples, 2 time point, there in one ChIP sample and one input sample in each time point.
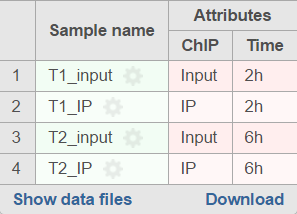 Figure 3. Experiment example data illustrate sample with two attributes: IP and Time
Figure 3. Experiment example data illustrate sample with two attributes: IP and Time
When select MACS2 task, the default option is to use sample attribute to add multiple pairs at one button click (Figure 4)
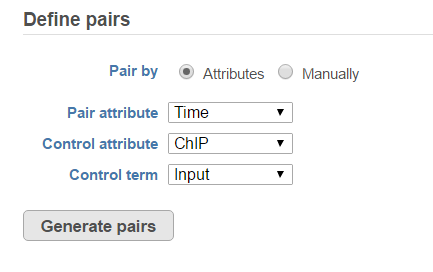 Figure 4. Specify IP vs control pairs based on sample attributes
Figure 4. Specify IP vs control pairs based on sample attributes
There are IP-Input pair in each time point, so the pair attribute is Time; Control attribute is the attribute contains IP and input group, which is ChIP, the control term is labeled as Input in the example, when click Generate pairs, the two pairs will be automatically added to the Pairs table at once (Figure 5).
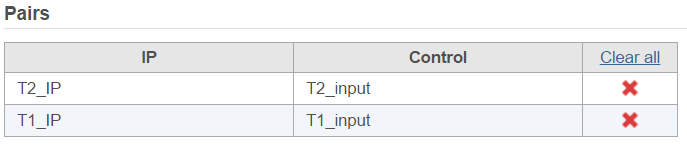 Figure 5. Two IP vs input sample pairs are added in the Pairs table
If multiple pairs are added in the Pairs table, the peak detection is performed on each pair independently.
Figure 5. Two IP vs input sample pairs are added in the Pairs table
If multiple pairs are added in the Pairs table, the peak detection is performed on each pair independently.
Peaks report
In the task report, each pair will generate a list of peaks displayed in a table (Figure 6)
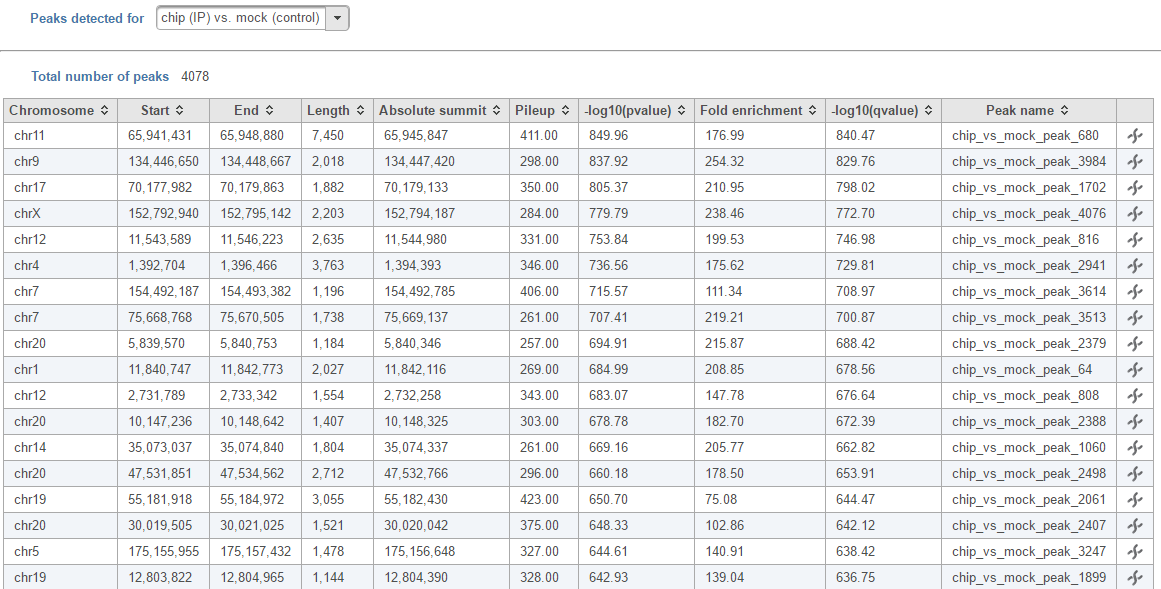 Figure 6. Peaks report on each IP vs control pair
In the report table, each row is a peak, besides genomic location of the peak region, it also include the following information:
Figure 6. Peaks report on each IP vs control pair
In the report table, each row is a peak, besides genomic location of the peak region, it also include the following information:
Absolute summit: base pair location of peak summit
Pileup: pileup height at peak summit
-log10(pvalue): negative log10 pvalue for thhe peak summit
Fold enricment: fold enrichhment for thhe peak summit against random Poisson distribution with local lambda
-log10(qvalue): negative log10 qvalue at peak summit
Click on the browse to peak button () to invoke chromosome view and zoom into that location. Click the Download button at the lower-right corner to download the peaks in a text file.
References
- Zhang Y, Liu T, et al. Model-based Analysis of ChIP-Seq (MACS). Genome Biol. 2008;9(9):R137.
Additional Assistance
If you need additional assistance, please visit our support page to submit a help ticket or find phone numbers for regional support.


| Your Rating: |
|
Results: |

|
1 | rates |
Overview
Content Tools