Variations in nucleotide sequence, in the form of single nucleotide variants (SNVs) and insertion and deletion events (INDELs), can exist within the germline or can be acquired by somatic alterations. Partek® Flow® provides pipeline creation tools to identify both SNVs and INDELs using aligned reads generated from targeted, exome, or whole genome DNA-Seq (or RNA-Seq) data. Detection of these variants can be performed by comparison against either the reference sequence utilized for alignment or among paired samples in a project. Tools for variant detection are performed on either Aligned reads or Filtered reads data nodes (Figure 1), and the Detect variants task will produce a Variants data node. The Variants data node will contain Variant Call Format (vcf) files for each sample in the project. Selecting a Variants data node will bring up the context sensitive menu, and the Download data option will allow for a zipped download of the vcf files in the project. Three detection tools are available under the Variant callers section of the context sensitive menu:
- Samtools
- FreeBayes
- LoFreq
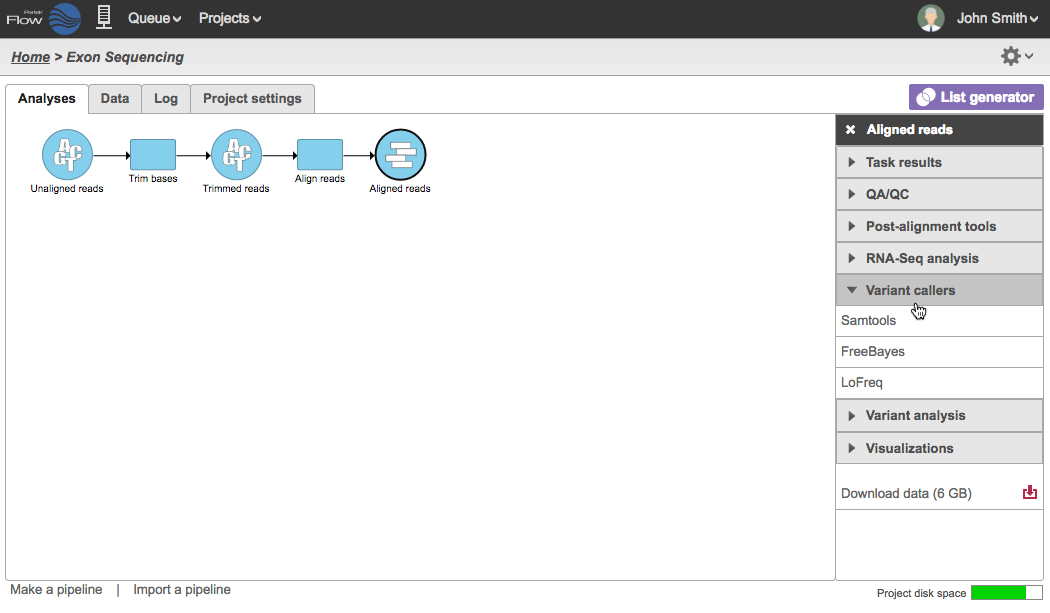 Figure 1. Showing Variant callers from an aligned reads node
Figure 1. Showing Variant callers from an aligned reads node
Additional Assistance
If you need additional assistance, please visit our support page to submit a help ticket or find phone numbers for regional support.


| Your Rating: |
|
Results: |

|
0 | rates |
Overview
Content Tools