Next, we will filter out certain cells and re-split the data. Re-splitting the data can be useful if you want to perform differential analysis and downstream analysis separately for proteins and genes. For your own analyses, re-splitting the data is optional. You could just as well continue with differential analysis with the merged data if you prefer.
Filter Groups
Because we have classified our cells, we can now filter based on those classifications. This can be used to focus on a single cell type for re-clustering and sub-classification or to exclude cells that are not of interest for downstream analysis.
- Click the Classified result data node
- Click Filtering
- Click Filter groups
- Set to exclude Cell type is Doublets using the drop-down menus
- Click AND
- Set the second filter to exclude Cell type is N/A using the drop-down menus
- Click Finish to apply the filter (Figure 1)
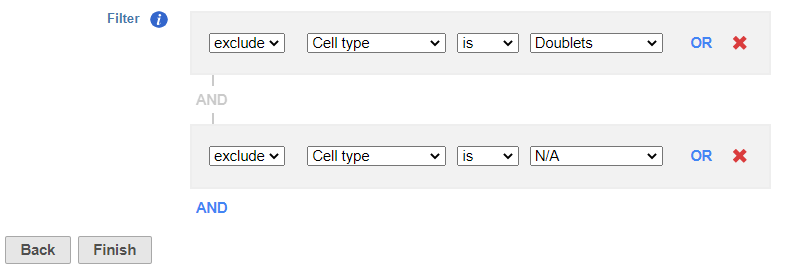 Figure 1. Set up the Filter groups task to exlcude Doublets and cells that are not classified
This produces a Filtered counts data node (Figure 2).
Figure 1. Set up the Filter groups task to exlcude Doublets and cells that are not classified
This produces a Filtered counts data node (Figure 2).
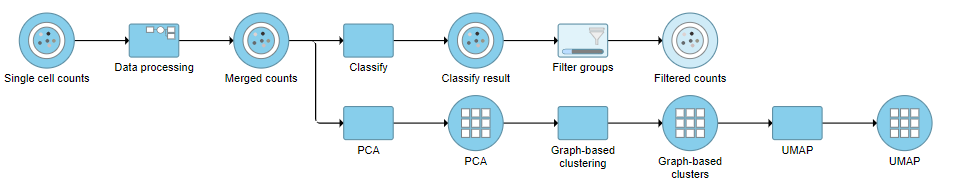 Figure 2. Filter groups output
Figure 2. Filter groups output
Re-split the Matrix
- Click the Filtered counts data node
- Click Pre-analysis tools
- Click Split by feature type
This will produce two data nodes, one for each data type (Figure 3). The split data nodes will both retain cell classification information.
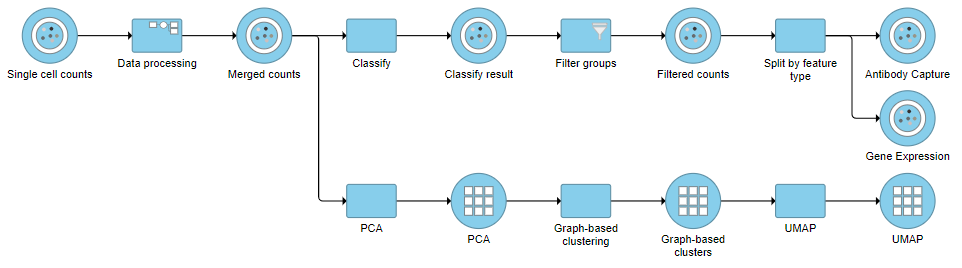 Figure 3. It is possible to re-split the merged matrix once again
Figure 3. It is possible to re-split the merged matrix once again
Differential Analysis and Visualization - Protein Data
Once we have classified our cells, we can use this information to perform comparisons between cell types or between experimental groups for a cell type. In this project, we only have a single sample, so we will compare cell types.
- Click the Antibody Capture data node
- Click Differential analysis
- Click GSA
The first step is to choose which attributes we want to consider in the statistical test.
- Check Cell type to include it in the statistical test
- Click Next
Next, we will set up the comparison we want to make. Here, we will compare the Activated and Mature B cells.
- Check Activated B cells in the top panel
- Check Mature B cells in the bottom panel
- Click Add comparison
The comparison should appear in the table as Activated B cells vs. Mature B cells.
- Click Finish to run the statistical test (Figure 4)
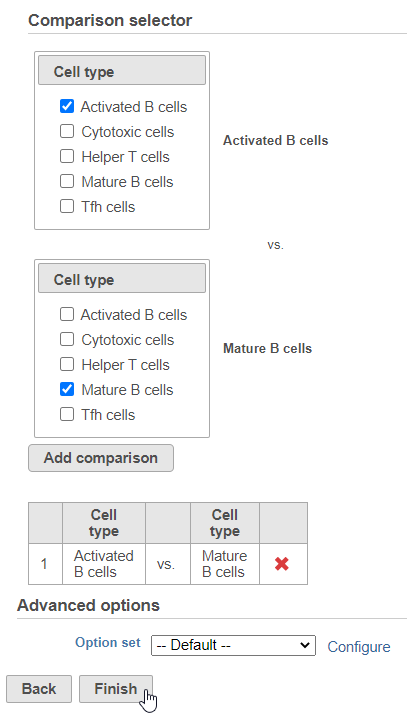 Figure 4. Setting up a comparison for differentially expressed proteins
The GSA task produces a GSA data node.
Figure 4. Setting up a comparison for differentially expressed proteins
The GSA task produces a GSA data node.
- Double-click the GSA data node to open the task report
The report lists each feature tested, giving p-value, false discovery rate adjusted p-value (FDR step up), and fold change values for each comparison (Figure 5).
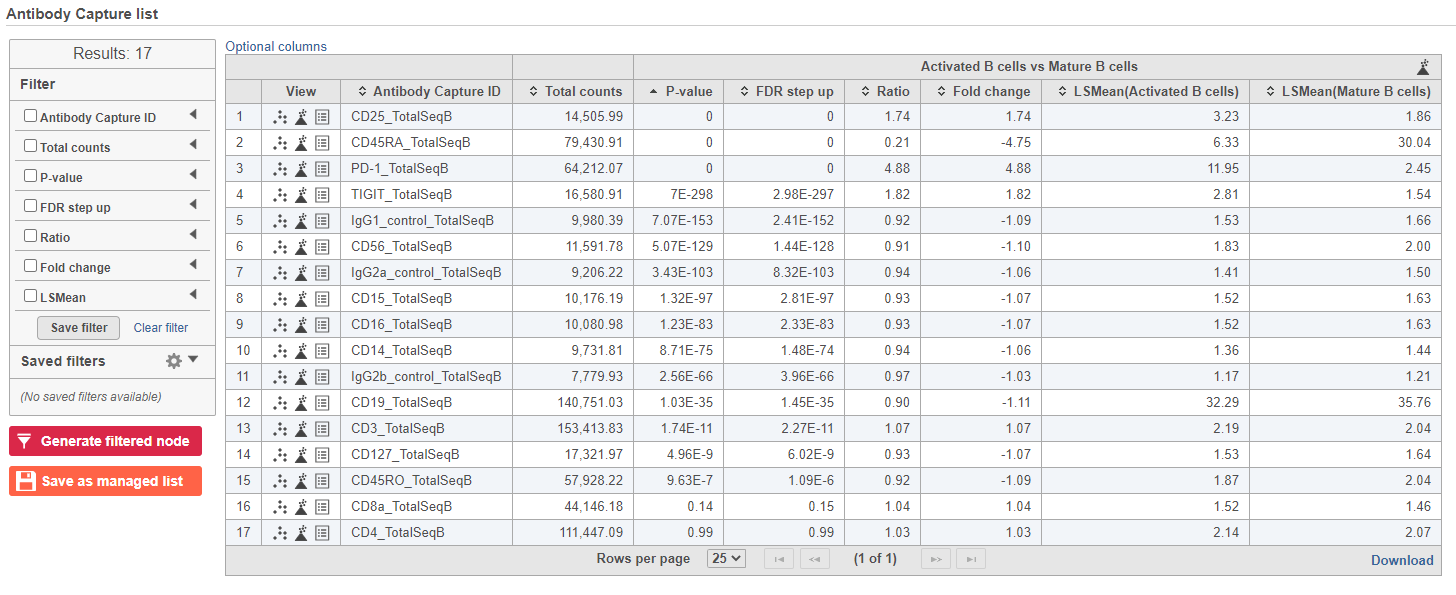 Figure 5. GSA report for protein expression data
In addition to the listed information, we can access dot and violin plots for each gene or protein from this table.
Figure 5. GSA report for protein expression data
In addition to the listed information, we can access dot and violin plots for each gene or protein from this table.
- Click
 in the CD45RA_TotalSeqB row
in the CD45RA_TotalSeqB row
This opens a dot plot in a new data viewer session, showing CD45A expression for cells in each of the classifications (Figure 6).
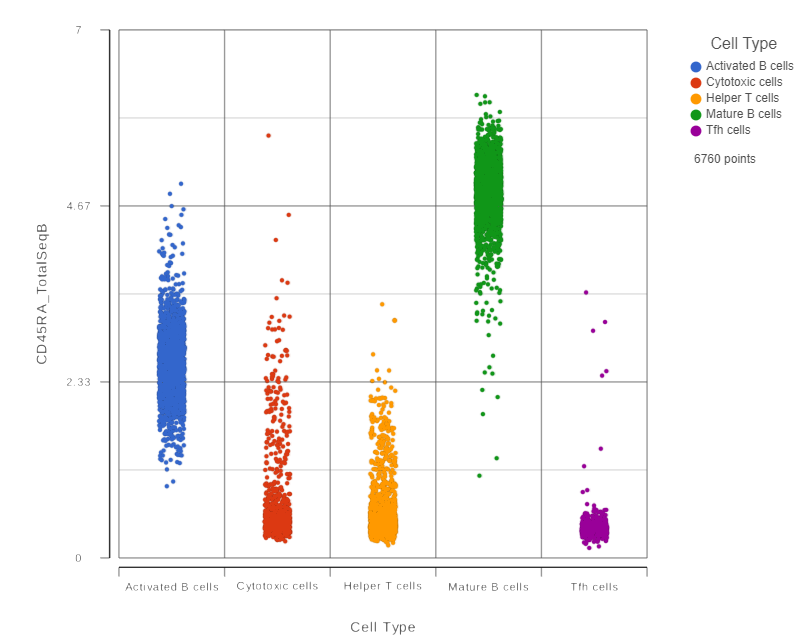 Figure 6. CD45RA dot plot for all cells
Figure 6. CD45RA dot plot for all cells
We can use the Configuration panel on the left to edit this plot.
- Open the Style icon
- Switch on Violins under Summary
- Switch on Overlay under Summary
- Switch on Colored under Summary
- Color by Graph-based clusters under Color and use the slider to decrease the Opacity
- Open the Axes icon
- Change the X axis data to Graph-based clusters
- Use the slider to increase the Jitter on the X axis (Figure 7)
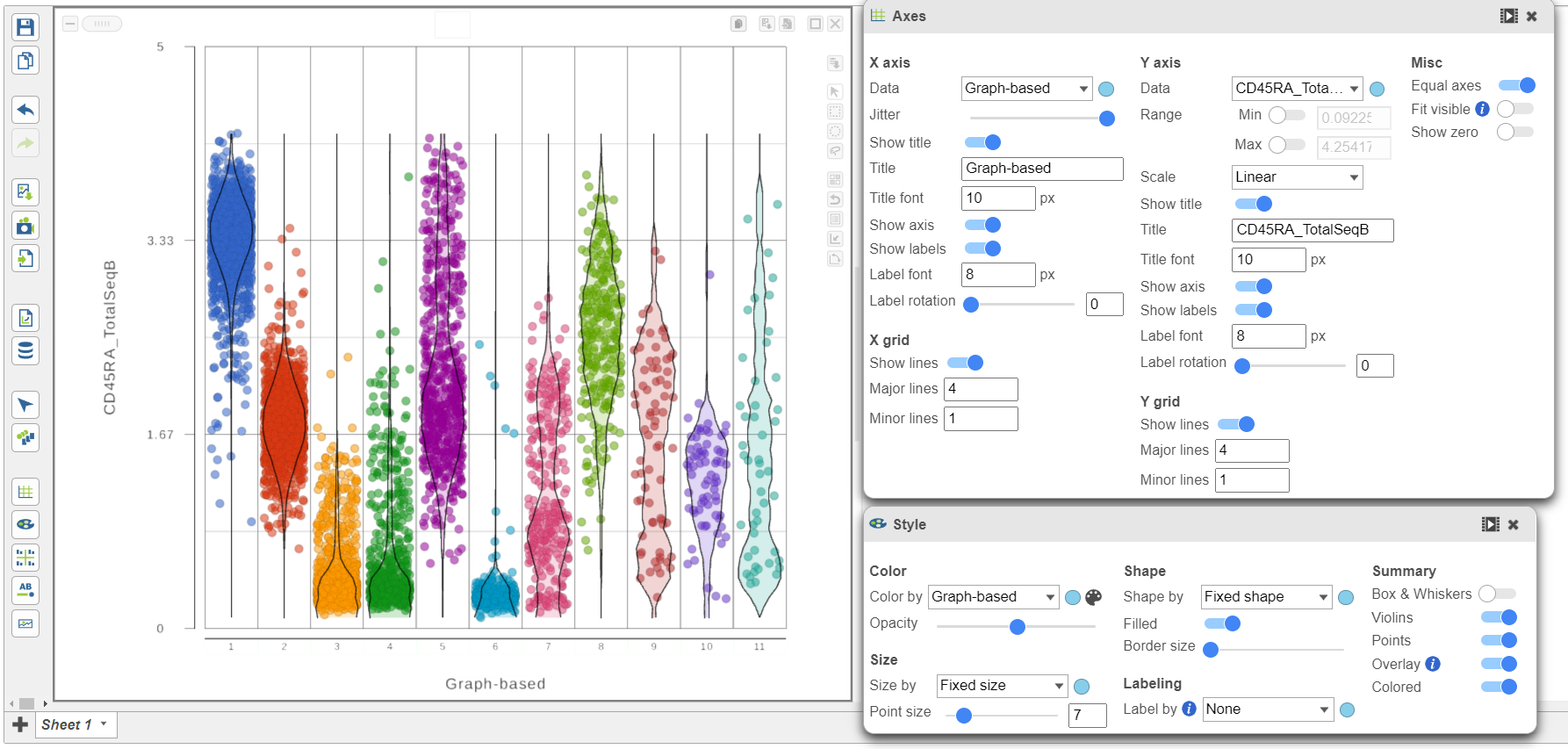 Figure 7. Configure the dot plot using the tools on the left
Figure 7. Configure the dot plot using the tools on the left
- Click the project name to return to the Analyses tab
To visualize all of the proteins at the same time, we can make a hierarchical clustering heat map.
- Click the GSA data node
- Click Exploratory analysis in the toolbox
- Click Hierarchical clustering/heatmap
- In the Ordering section, choose Graph-based clusters from the Cell order drop-down list
- Click Finish to run with the other default settings
- Double-click the Hierarchical clustering task node to open the heatmap
The heatmap can easily be customized using the tools on the left.
- Open the Axes icon
- Switch off Show Row labels
- Increase the Font to 16 (Figure 8)
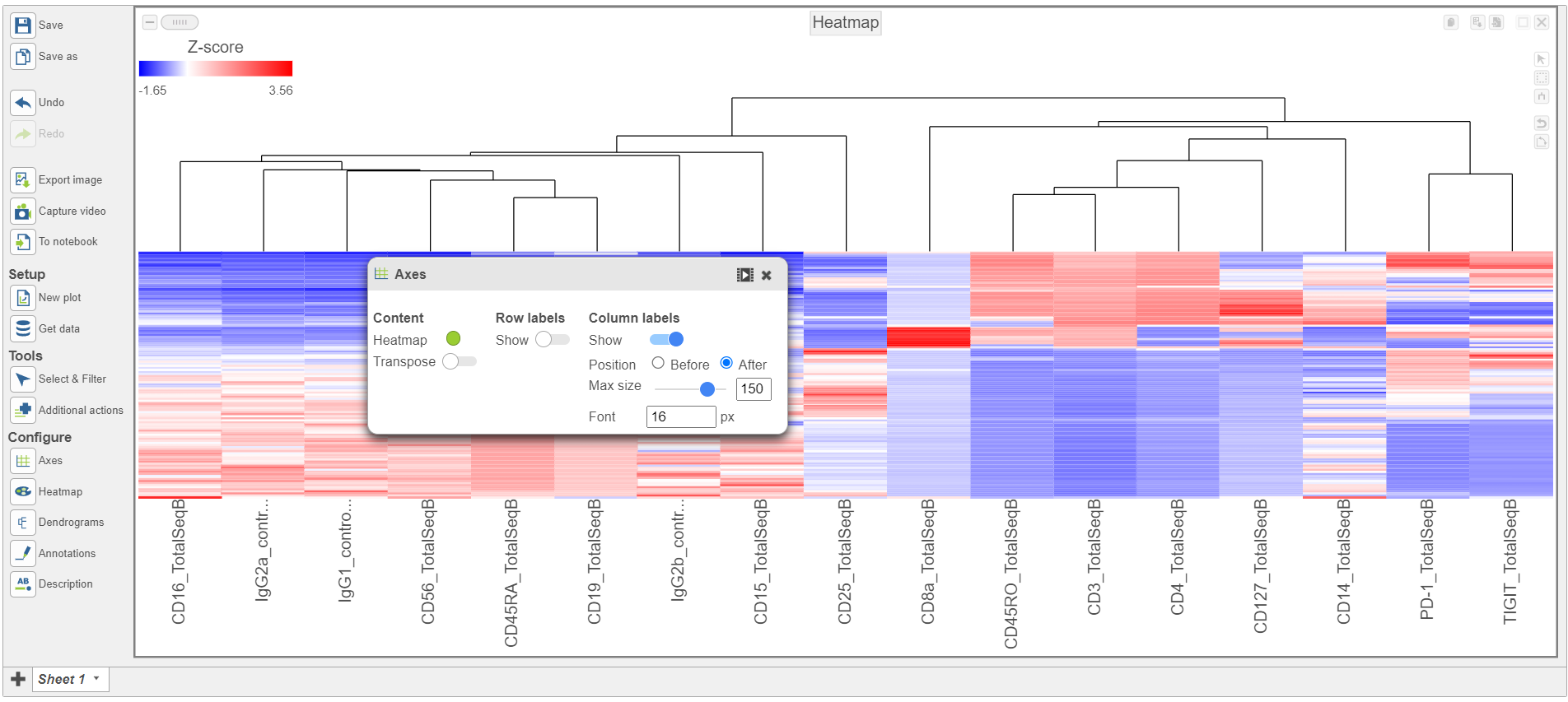 Figure 8. Heatmap showing altered Axes labels
Figure 8. Heatmap showing altered Axes labels
- Activate the Transpose switch which will switch the Row and Column labels, so now the Row labels will be shown (Figure 9)
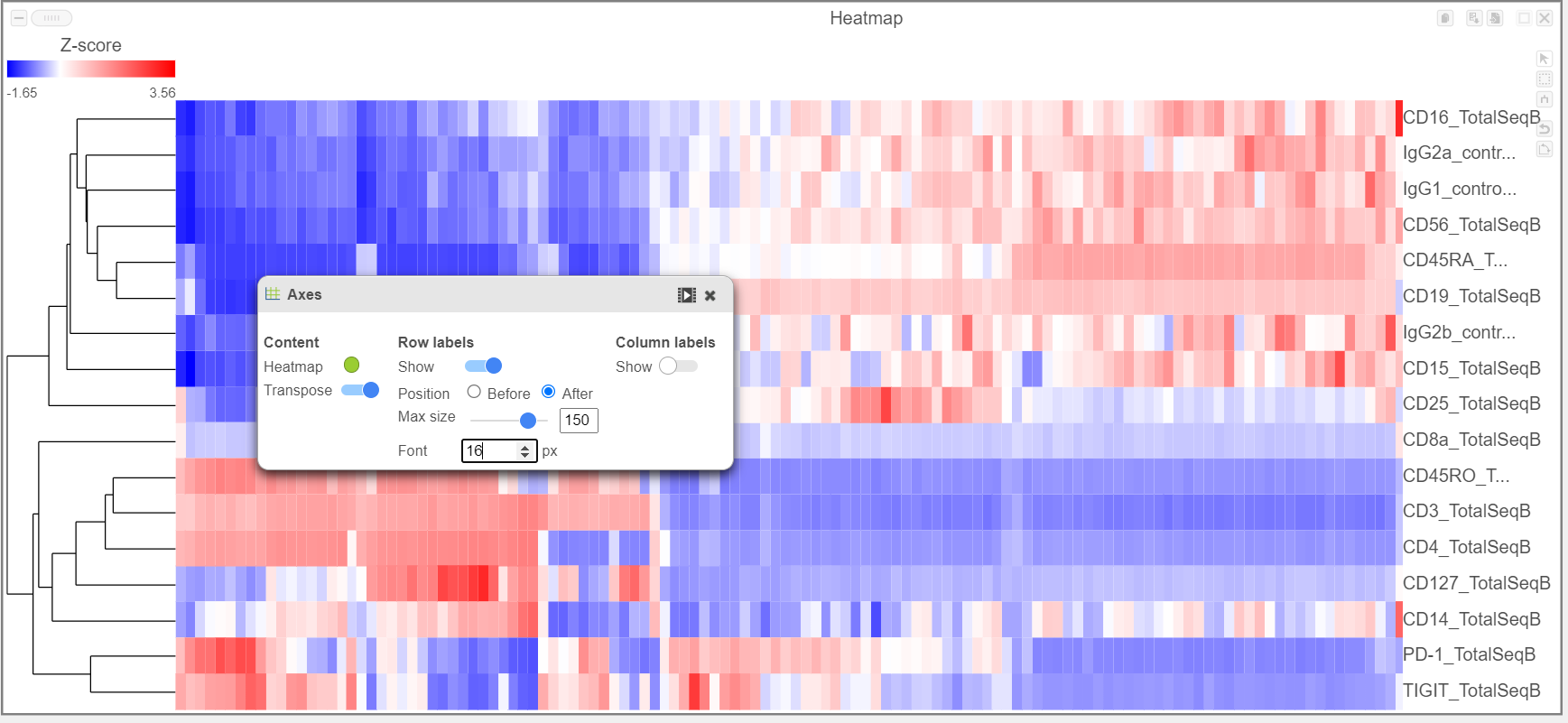 Figure 9. Transpose the Heatmap to switch the columns and rows
Figure 9. Transpose the Heatmap to switch the columns and rows
- Open the Dendrograms icon
- Choose Row color By cluster and change Row clusters to 4
- Change Row dendrogram size to 80 (Figure 10)
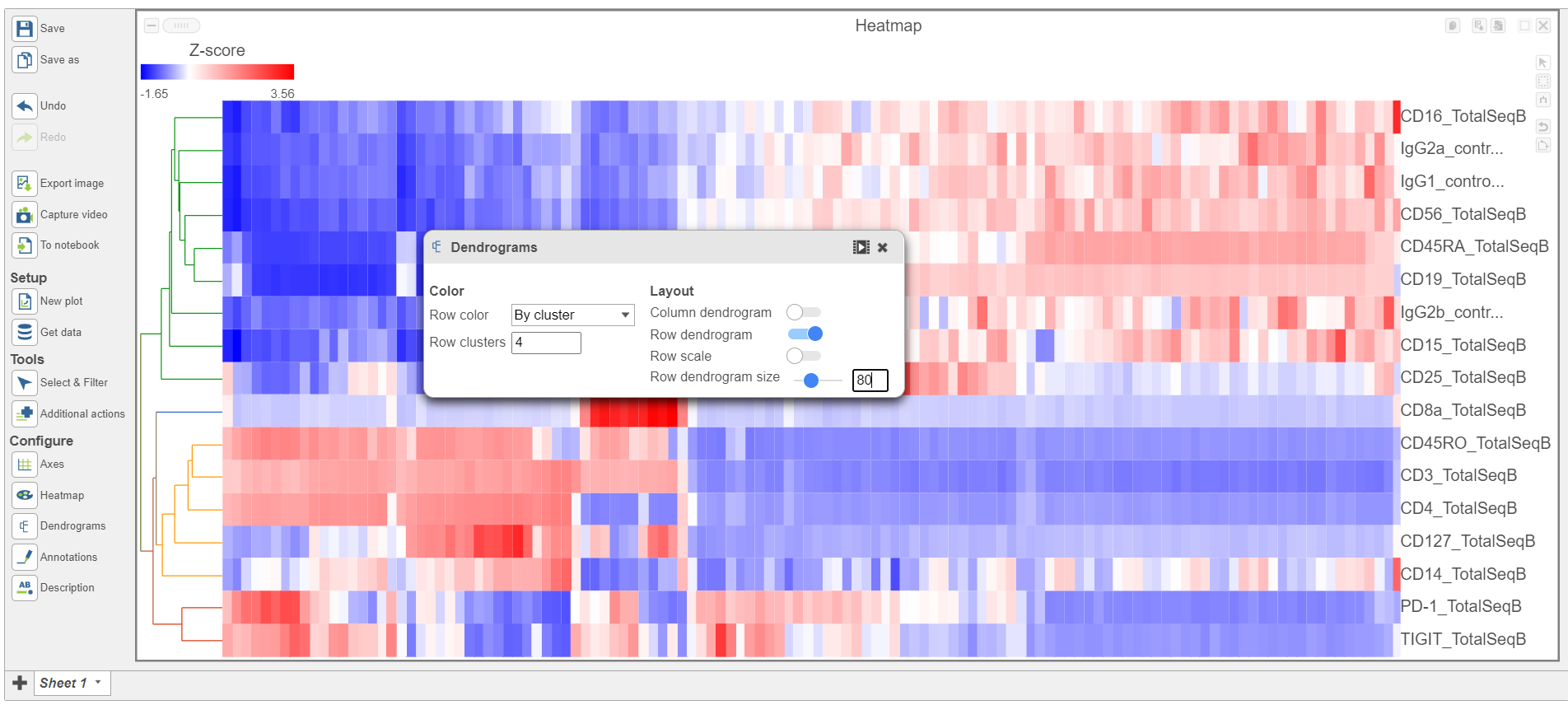 Figure 10. Configure the Dendrograms settings
Figure 10. Configure the Dendrograms settings
- In the Heatmap icon
- Navigate to Range under Color
- Set the Min and Max to -1.2 and 1.2, respectively
- Change the Shape to Circle (Figure 11)
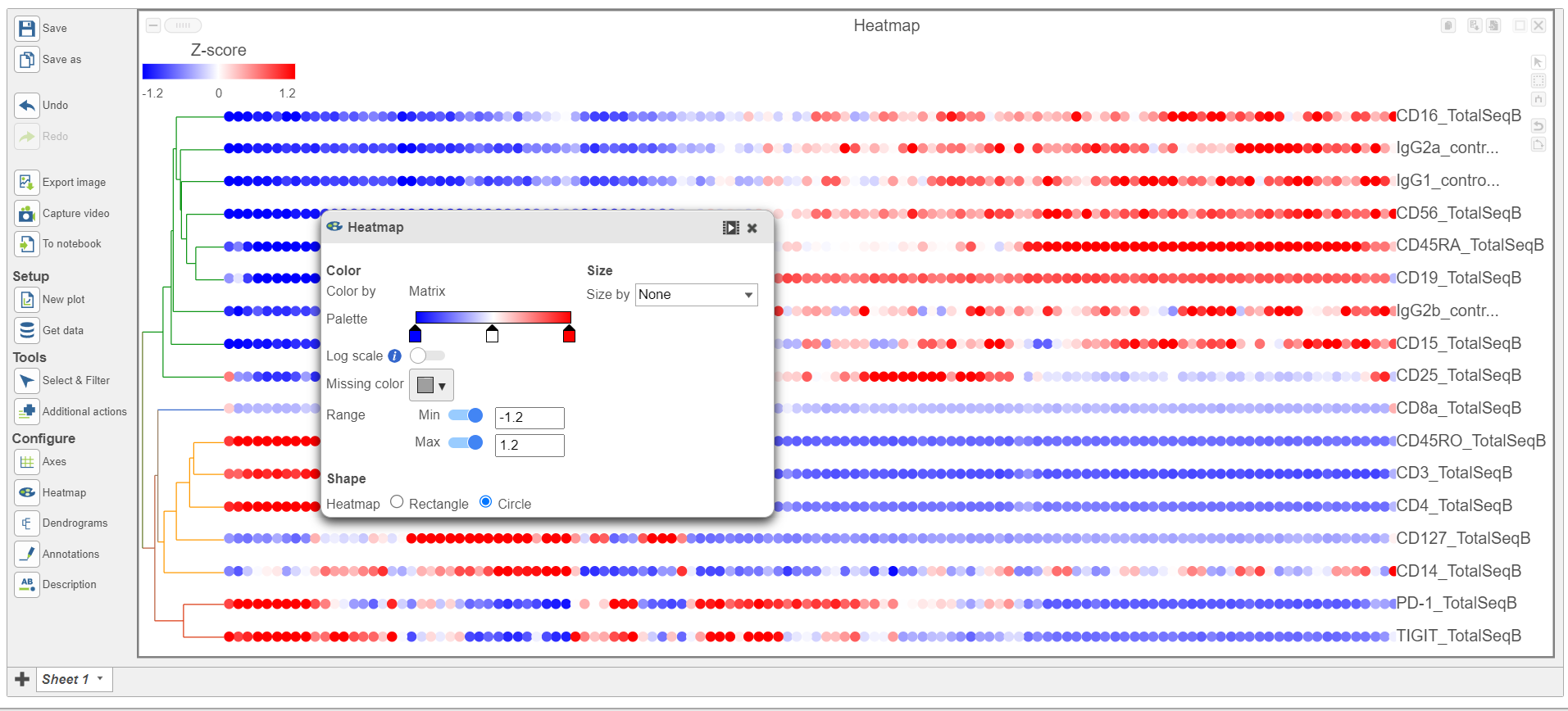 Figure 11. Configure the Heatmap icon
Figure 11. Configure the Heatmap icon
- Switch the Shape back to Rectangle
- Change the Color Palette by clicking on the color squares and selecting colors from the rainbow. Click outside of the selection box to exit this selection. The color options can be dragged alone the Palette to highlight value differences (Figure 12).
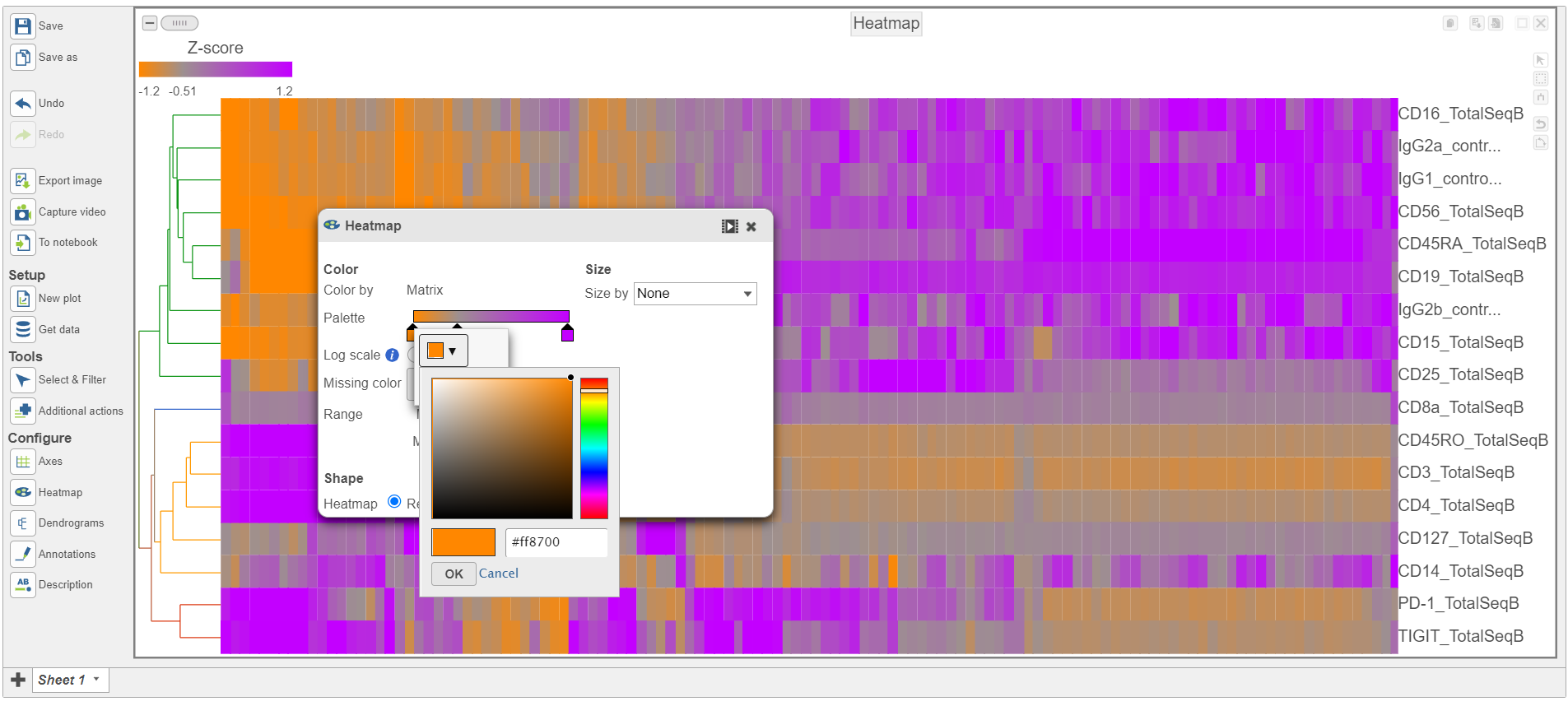 Figure 12. Heatmap showing expression of protein markers after changing the Heatmap settings further
Feel free to explore the other tool options on the left to customize the plot further.
Figure 12. Heatmap showing expression of protein markers after changing the Heatmap settings further
Feel free to explore the other tool options on the left to customize the plot further.
Differential Analysis, Visualization, and Pathway analysis - Gene Expression Data
We can use a similar approach to analyze the gene expression data.
- Click the project name to return to the Analyses tab
- Click the Gene Expression data node
- Click Differential analysis
- Click GSA
- Check Cell type to include it in the statistical test
- Click Next
- Check Activated B cells in the top panel
- Check Mature B cells in the bottom panel
- Click Add comparison
- Click Finish to run the statistical test
As before, this will generate a GSA task node and a GSA data node.
- Double-click the GSA task node to open the task report (Figure 13)
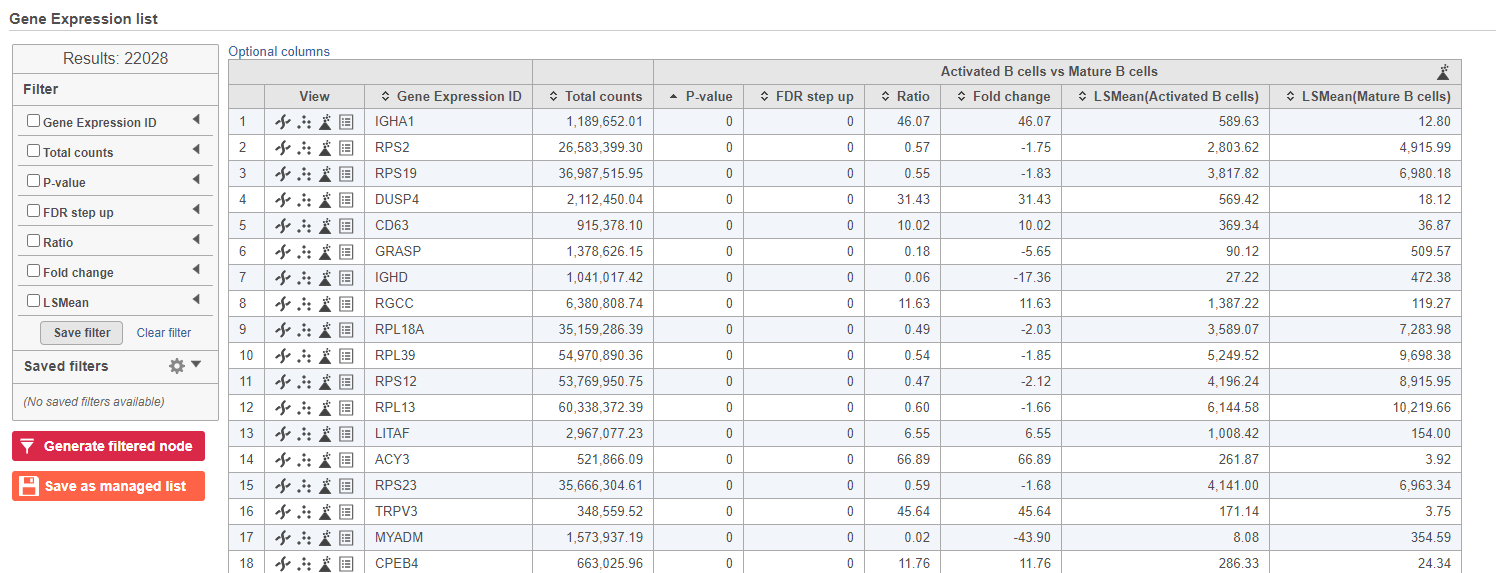 Figure 13. GSA report for the gene expression data
Because more than 20,000 genes have been analyzed, it is useful to use a volcano plot to get an idea about the overall changes.
Figure 13. GSA report for the gene expression data
Because more than 20,000 genes have been analyzed, it is useful to use a volcano plot to get an idea about the overall changes.
- Click
 in the top right corner of the table to open a volcano plot
in the top right corner of the table to open a volcano plot
The Volcano plot opens in a new data viewer session, in a new tab in the web browser. It shows each gene as a point with cutoff lines set for P-value (y-axis) and fold-change (x-axis). By default, the P-value cutoff is set to 0.05 and the fold-change cutoff is set at |2| (Figure 14).
The plot can be configured using various options in the Configuration card on the left. For example, the Color, Size and Shape cards can be used to change the appearance of the points. The X and Y-axes can be changed in the Data card. The Significance card can be used to set different Fold-change and P-value thresholds for coloring up/down-regulated genes.
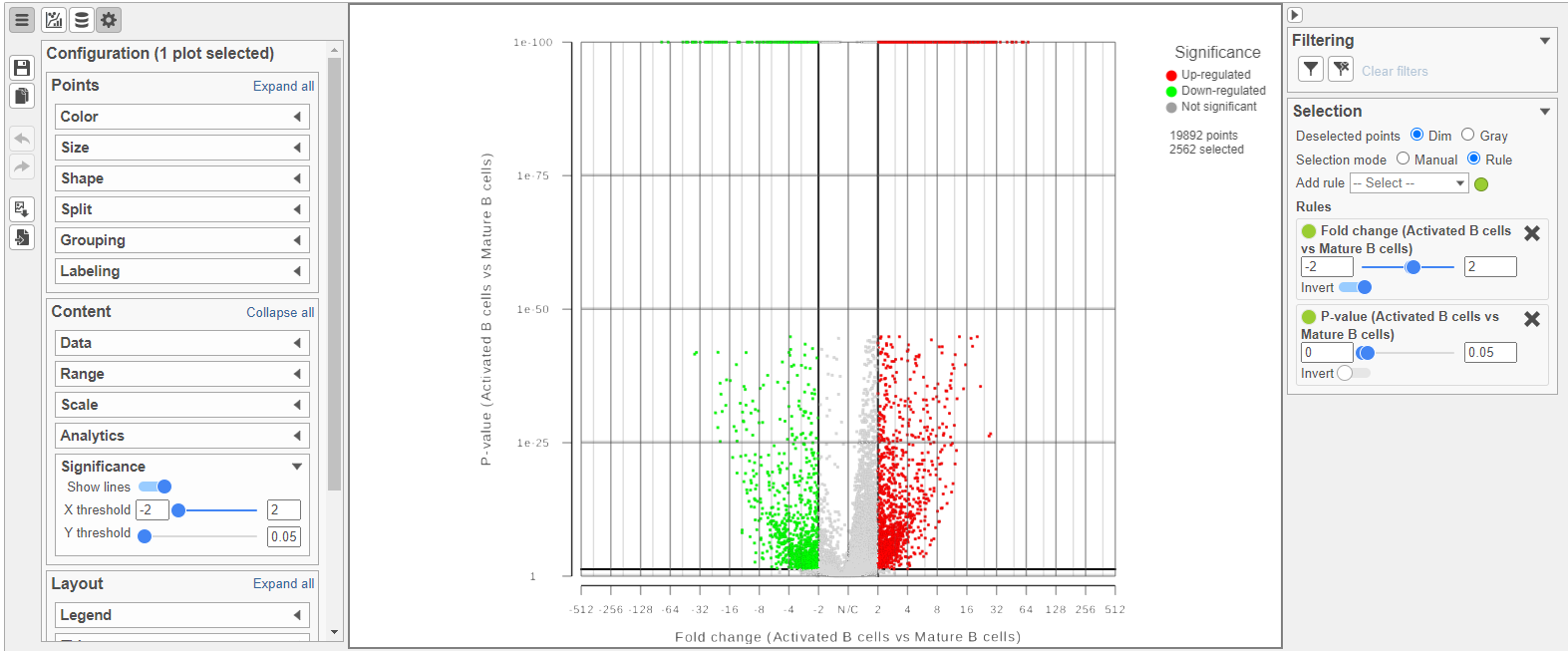 Figure 14. The volcano plot can be configured using various options in the Configuration and Selection cards
Figure 14. The volcano plot can be configured using various options in the Configuration and Selection cards
- Click the GSA report tab in your web browser to return to the full report
We can filter the full set of genes to include only the significantly different genes using the filter panel on the left.
- Click FDR step up
- Type 0.05 for the cutoff and press Enter on your keyboard
- Click Fold change
- Set to From -2 to 2 and press Enter on your keyboard
The number at the top of the filter will update to show the number of included genes (Figure 15).
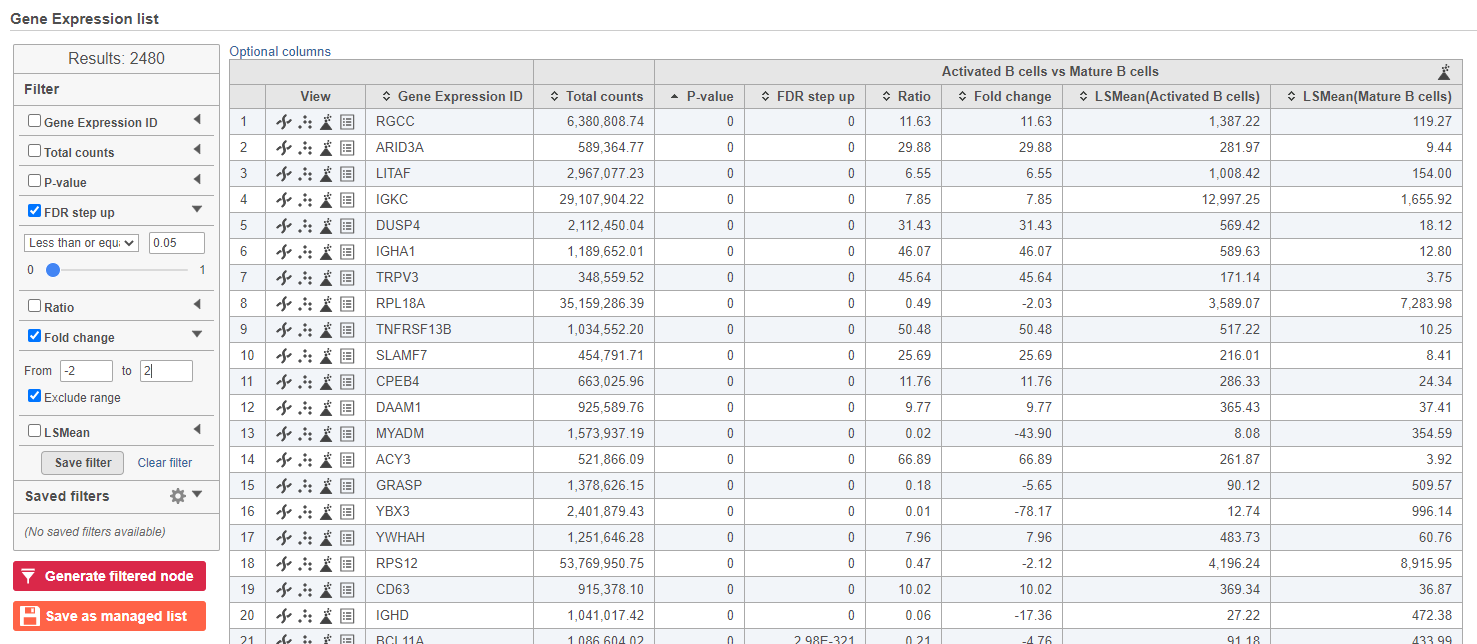 Figure 15. Use the panel on the left to filter the list for significant genes
Figure 15. Use the panel on the left to filter the list for significant genes
- Click
to create a new data node including only these significantly different genes
A task, Differential analysis filter, will run and generate a new Filtered Feature list data node. We can get a better idea about the biology underlying these gene expression changes using gene set or pathway enrichment. Note, you need to have the Pathway toolkit enabled to perform the next steps.
- Click the Filtered feature list data node
- Click Biological interpretation in the toolbox
- Click Pathway enrichment
- Make sure that Homo sapiens is selected in the Species drop-down menu
- Click Finish to run
- Double-click the Pathway enrichment task node to open the task report
The pathway enrichment results list KEGG pathways, giving an enrichment score and p-value for each (Figure 16).
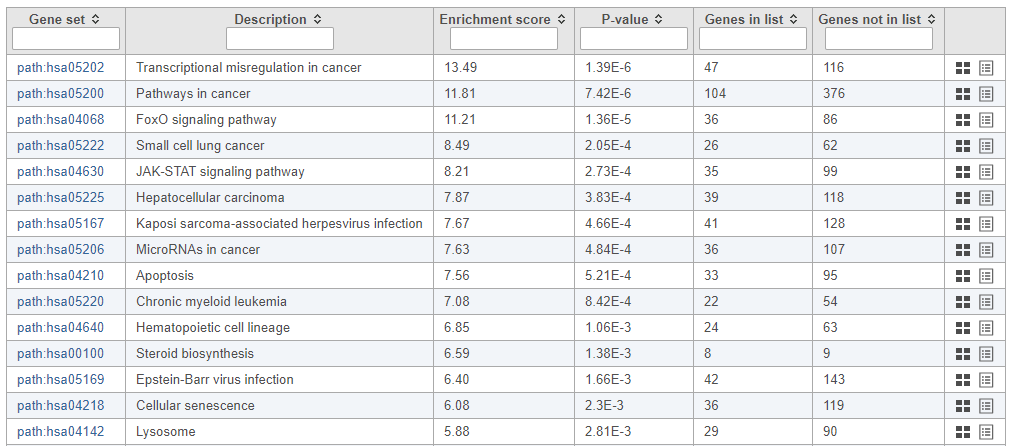 Figure 16. Results of pathway enrichment test
To get a better idea about the changes in each enriched pathway, we can view an interactive KEGG pathway map.
Figure 16. Results of pathway enrichment test
To get a better idea about the changes in each enriched pathway, we can view an interactive KEGG pathway map.
- Click path:hsa05202 in the Transcriptional misregulation in cancer row
The KEGG pathway map shows up-regulated genes from the input list in red and down-regulated genes from the input list in green (Figure 17).
![]() Figure 17. Transcriptional misregulation in cancer pathway with significant genes highlighted in green and red
Figure 17. Transcriptional misregulation in cancer pathway with significant genes highlighted in green and red
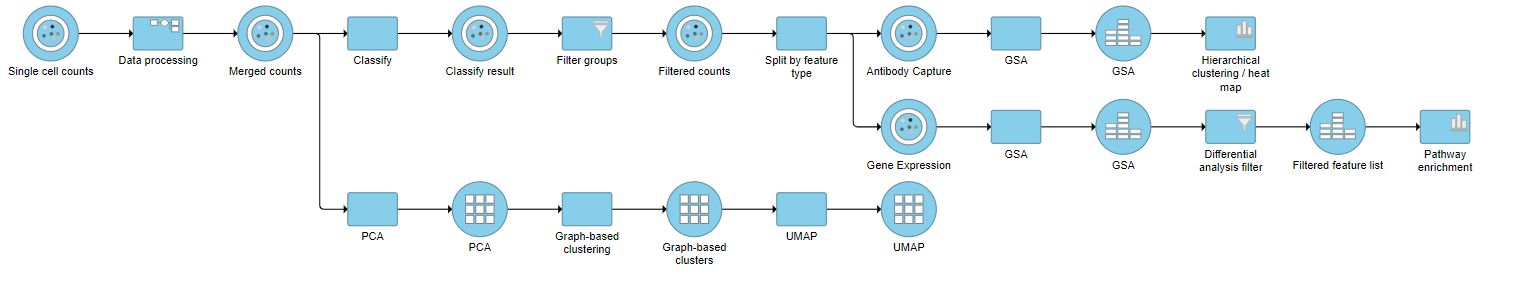 Figure 18. Final CITE-Seq pipeline
Figure 18. Final CITE-Seq pipeline
Additional Assistance
If you need additional assistance, please visit our support page to submit a help ticket or find phone numbers for regional support.


| Your Rating: |
|
Results: |

|
2 | rates |
Overview
Content Tools