Hierarchical clustering is a statistical method used to assign similar objects into groups called clusters. It is typically performed on results of statistical analyses, such as a list of significant genes/transcripts, but can also be invoked on the full data set, as a part of exploratory analysis.
Hierarchical clustering is an unsupervised technique, meaning that the number of clusters is not specified up front. In the beginning, each row and/or column is considered a cluster. The two most similar clusters are combined and continue to combine until all objects are in the same cluster. Hierarchical clustering produces a tree (called a dendrogram) that shows the hierarchy of the clusters.
This tutorial will illustrate how to:
Invoking Hierarchical Clustering
To invoke hierarchical clustering, select a data node containing count data (e.g. Gene counts, Normalized counts, Single cell counts), or a Feature list data node (to cluster significant genes/transcripts) and then click on the Hierarchical clustering / heat map option in the context sensitive menu (Figure 1).
Figure 1. Hierarchical clustering as a part of visualisation tools
The hierarchical clustering setup dialog (Figure 2) enables you to control the clustering algorithm. Starting from the top, you can choose to Cluster samples, Cluster features (genes/transcripts) or both. By default, if there are less than 3000 samples, the Cluster samples check button is selected. Otherwise the check button is de-selected. If Cluster samples is unchecked, the Ordering option becomes active (see below).
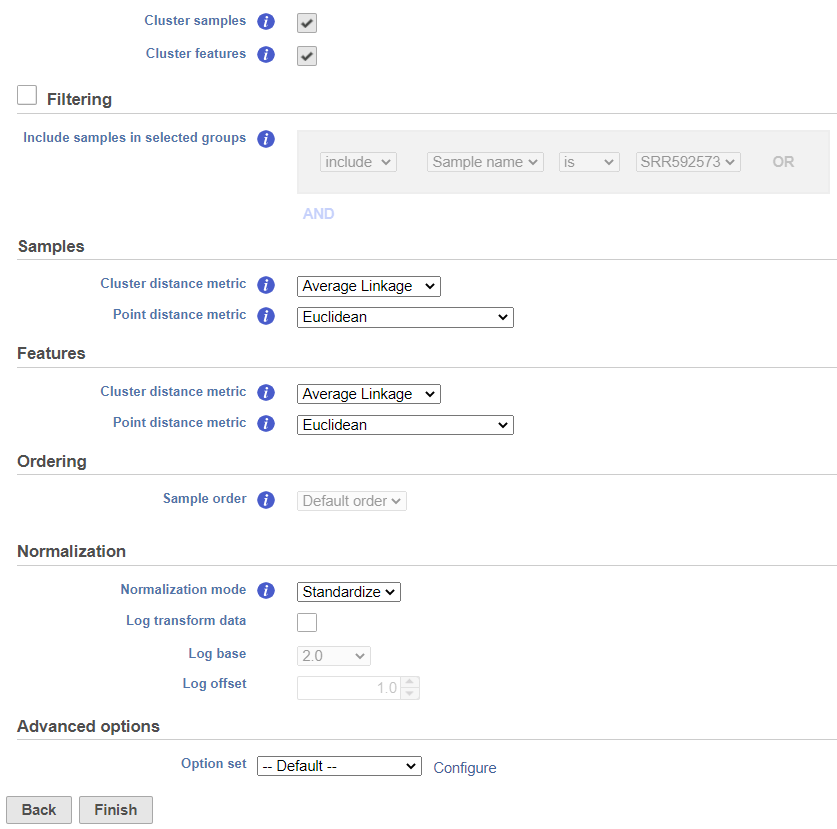
Figure 2. Setup dialog of hierarchical clustering (default settings)
If you do not want to cluster all the samples, but select a subset based on a specific sample or cell attribute (i.e. group membership), check the Filtering option and set a filtering rule using the drop down lists (Figure 3). The default value of the Filtering option is All samples.
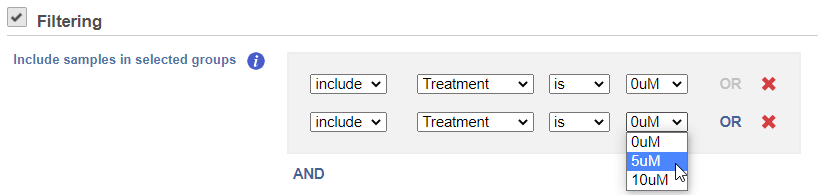
Figure 3. Specifying a subset of data for clustering, based on sample attributes. In the example on the figure, only the samples belonging to the 0uM and 5uM groups will be clustered. Samples belonging to other treatment groups will be omitted.
Cluster distance metric for samples and features is used to determine how the distance between two clusters will be calculated (Figure 4):
- Single Linkage: the distance between two clusters is determined by the distance of the closest objects in the two clusters
- Complete Linkage: the distance between two clusters is equal to the distance between the two furthest members of those clusters
- Average Linkage: the average distance between all the pairs of objects in the two different clusters is used as the measure of distance between the two clusters
- Centroid method: the distance between two clusters is equal to the distance between the centroids of those clusters
- Ward's method: the distance between two clusters is designed to minimize the size of an error measure based on the sum of squares
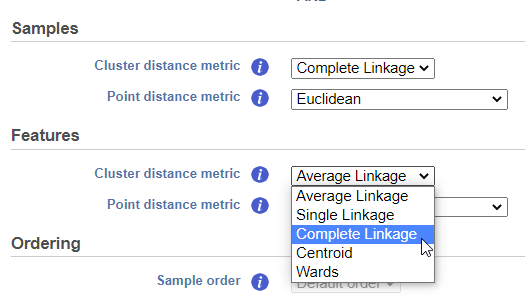 Figure 4. The cluster distance metric can be chosen for samples and features
Figure 4. The cluster distance metric can be chosen for samples and features
Point distance metric is used to determine the distance between two rows or columns. For more detailed information about the equations, we refer you to the distance metrics chapter.
If the Cluster samples box is unchecked, the Ordering option becomes active (Figure 5). Choose an attribute from the drop down list. Click and drag to rearrange the order of groups.
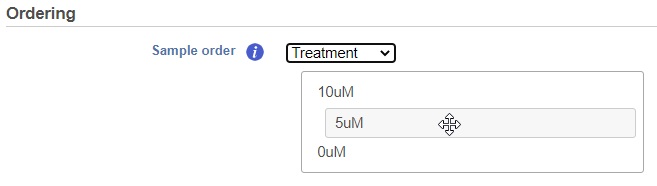 Figure 5. The order of samples in the heatmap can be customized
You can choose how the data is normalized. Under the Normalization mode dropdown, Standardize (default) will make each column mean as zero and standard deviation as 1 in all features. This is the default normalization and it makes all the features (e.g., genes) have equal weight. Standardized values are also known as Z-scores. The normalization mode Shift will make each column mean as zero. Choose None to perform clustering on the values in the quantified data node. The data can also be Log2 transformed on the fly.
Figure 5. The order of samples in the heatmap can be customized
You can choose how the data is normalized. Under the Normalization mode dropdown, Standardize (default) will make each column mean as zero and standard deviation as 1 in all features. This is the default normalization and it makes all the features (e.g., genes) have equal weight. Standardized values are also known as Z-scores. The normalization mode Shift will make each column mean as zero. Choose None to perform clustering on the values in the quantified data node. The data can also be Log2 transformed on the fly.
Another way to invoke heatmap without performing clustering is in the data viewer. When you select the Heatmap ![]() icon in the available plots, data nodes that contains two dimension matrix can be use to draw this type of plot.
icon in the available plots, data nodes that contains two dimension matrix can be use to draw this type of plot.
Heat Map
The output of a Hierarchical clustering task is a heat map (Figure 6) with or without dendrogram depends on whether you perform cluster on samples/cells or features. By default, samples are on rows (sample labels are displayed as seen in the Data tab) and features (genes or transcripts, depending on the input data) on columns. Colors are based on standardized expression values (default selection; performed on the fly). Dendrograms show clustering of rows (samples) and columns (variables).
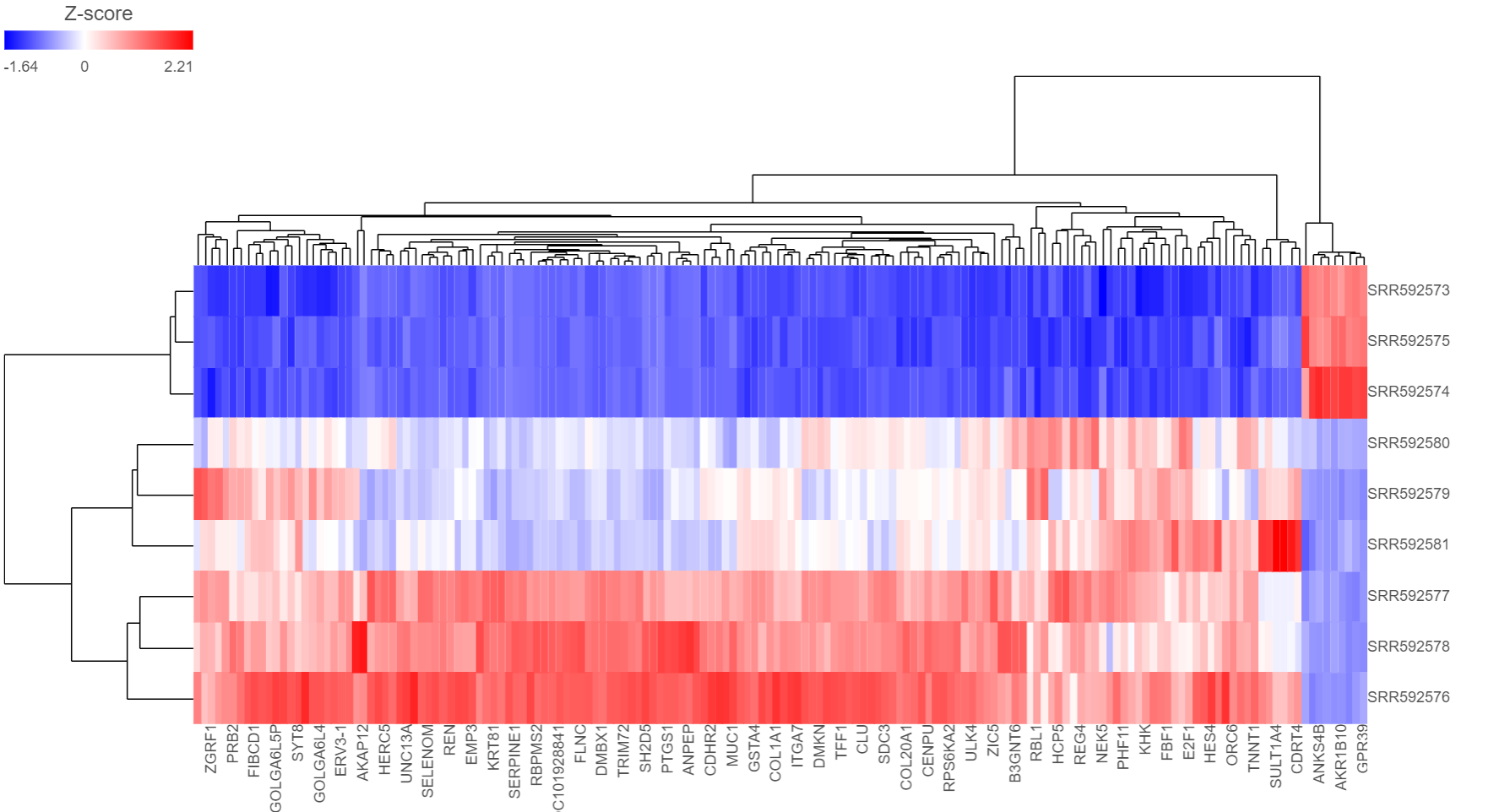
Figure 6. Heat map. Samples are on columns, variables (in this example: genes) on columns, and the heat map is based on standardised gene expression values
Depending on the resolution of your screen and the number of samples and variables that need to be displayed, some binning may be involved. If there are more than samples/genes than pixels, values of neighboring components will be averaged together. When you zoom in to certain level, you will see each cell represent one sample/gene. To zoom, use the mouse wheel to zoom in / out. To move the map around when zoomed in, press down the right button of the mouse and drag the map.
There are 5 sections in the Configuration panel (Figure 7): Content, Heatmap, Dendrograms, Annotations and Layout. Click on the triangle (![]() ) to expand the section to configure.
) to expand the section to configure.
Figure 7. Heat map controls
Content
This section controls the data source used to draw the values in the heatmap. The heatmap is a color representation of the values in the matrix selected. In addition to color, you can also use the Size drop down list to size by a set of values. Most of the data nodes contain only one matrix, so the only options available in the Size drop down are None or Matrix (Figure 8).
Figure 8. When data node contains only one matrix, not need to use size by configuration
However, if a data node contains multiple matrix information, e.g. if you perform descriptive statistics on cluster groups for every gene like mean, std. dev, percent of detected cells, etc, each stats result will be in a separate matrix in the output data node. You might want to use color of the component in the heatmap to represent one type of stats (like mean of the groups) and size of the component to represent the information from a different statistic (like std. dev) (Figure 9).
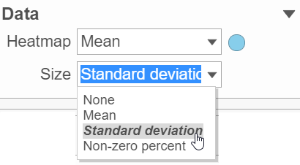
Figure 9. When a data node containing more than one matrices, different matrices can be used to color and size the components
Heatmap
This section is used to configure the color and shape of the components in the heatmap (Figure 10).
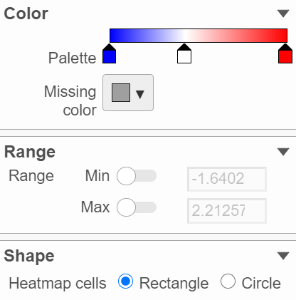
Figure 10. Configure heatmap color and shape
In the color palette horizontal bar, the left side color represents the lowest value and the right side color represents the highest value in the matrix data represented. By default, there are 3 tabs (![]() ): minimum, middle, and maximum color value of the default range calculated on the matrix. Left-click on the middle tab and drag left/right can change the middle the value this tab represents. If you left-click on the middle tab once, you can change the color and value this tab represents (Figure 11). Click on (
): minimum, middle, and maximum color value of the default range calculated on the matrix. Left-click on the middle tab and drag left/right can change the middle the value this tab represents. If you left-click on the middle tab once, you can change the color and value this tab represents (Figure 11). Click on (![]() ) to remove this tab.
) to remove this tab.
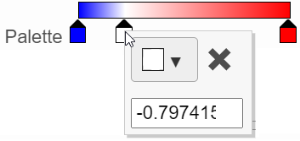
Figure 11. Left click on the tab to change the color and value represented
Click on the little triangle next to the color square (![]() ) to choose a color to represent the value by clicking on a color or type in the RGB color of the color, click OK (Figure 12).
) to choose a color to represent the value by clicking on a color or type in the RGB color of the color, click OK (Figure 12).
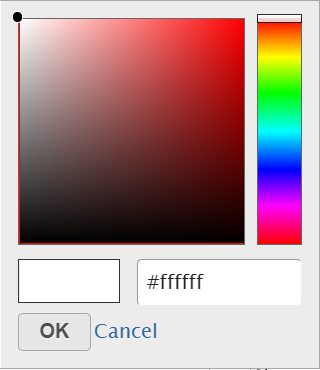
Figure 12. Select a color from the color palette
The min and max tabs cannot be dragged or removed. If you left-click on them, you can choose a different color. When you click on the Palette bar, you can add a new color tab between min and max (Figure 13). Adding a tab can be useful when there is an outlier value in the data. You can use a different color to represent different value range.
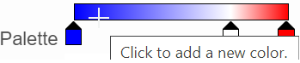
Figure 13. Add a new color tab between min and max
To change the min and max threshold values represented by the color palette, click on the toggle switch (![]() ) in the Range card, and specify the values in the text boxes.
) in the Range card, and specify the values in the text boxes.
The shape of the heatmap cell (component) can be configured either as a rectangle or circle by selecting the radio button in the shape section.
Dendrograms
If cluster analysis is performed on samples and/or features, the result will be displayed in dendrograms. By default, the dendrograms are all colored in black (Figure 6).
The color of the dendrograms can be configured (Figure 14.)
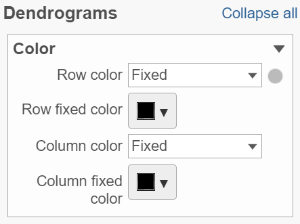
Figure 14. Configure the color of dendrograms
Click on the color square triangle (![]() ) to choose a different color for the dendrogram.
) to choose a different color for the dendrogram.
When the By cluster in the Row/Column color drop-down list, the number of clusters needs to be specified (Figure 15). The top N number of clusters will be in N different colors.
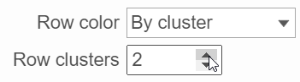
Figure 15. Configure the dendrogram color based on number of cluster specified
Annotations
This section allows you to add sample or cell level annotation to the viewer. First, make sure to choose the correct data node which contains the annotation information you would like to use by clicking the circle (![]() ). All project level annotation will be available on all data nodes in the pipeline (Figure 16). Choose an attribute from the Row annot drop-down list. Multiple attributes can be chosen from the drop-down list and can be reordered by clicking and dragging the groups below the drop-down list.
). All project level annotation will be available on all data nodes in the pipeline (Figure 16). Choose an attribute from the Row annot drop-down list. Multiple attributes can be chosen from the drop-down list and can be reordered by clicking and dragging the groups below the drop-down list.
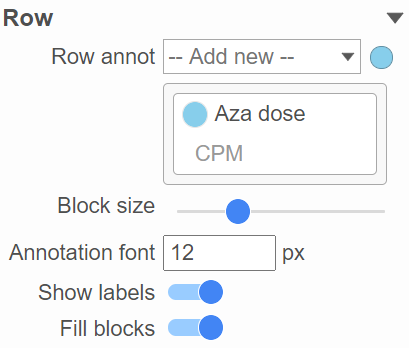
Figure 16. Configure sample or cell annotation
Each attribute is represented as an annotation bar next to the heatmap. Different colors represent the different groups in the attribute. The width of the bar can be adjusted by Block size slider when the Show labels toggle switch is on. The text of the label font size can be changed by specifying pixel size. The Fill blocks toggle switch adds or removes color from the annotation labels.
Layout
To change the orientation of the plot, click on the toggle switch of Data ( ) in the Layout section (Figure 17).
) in the Layout section (Figure 17).
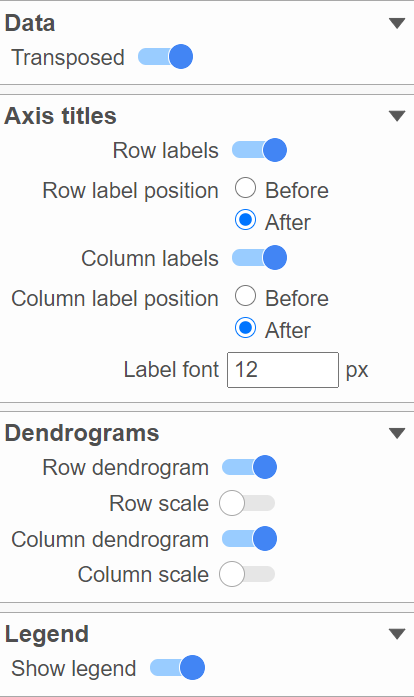
Figure 17. Configure the layout of the plot
Axis titles, dendrograms, and the legend can be turned on or off by clicking the relevant toggle switches. The axes label font size can be changed by specifying the number of pixels.
In-plot controls
In the mode bar of the viewer (in the top right corner of the heatmap), there is a Flip mode button (![]() ). In flip mode, you can click on a line in the dendrogram which represents a cluster branch and the location of the two legs of the branch will be swapped.
). In flip mode, you can click on a line in the dendrogram which represents a cluster branch and the location of the two legs of the branch will be swapped.
In move mode (![]() ), you can left-click and drag to move around the heatmap if you are zoomed in. Left-clicking once on the heatmap or on a dendrogram branch will select that rows/column.
), you can left-click and drag to move around the heatmap if you are zoomed in. Left-clicking once on the heatmap or on a dendrogram branch will select that rows/column.
In selection mode (![]() ), you can click and drag to select a range of rows, columns, or components.
), you can click and drag to select a range of rows, columns, or components.
Additional Assistance
If you need additional assistance, please visit our support page to submit a help ticket or find phone numbers for regional support.


| Your Rating: |
|
Results: |

|
4 | rates |
Overview
Content Tools