We will now examine the results of our exploratory analysis and use a combination of techniques to classify different subsets of T and B cells in the MALT sample.
Exploratory Analysis Results
- Double click the UMAP data node
- In the Configuration card on the left, expand the Color card and color the cells by the Graph-based attribute (Figure ?)
 Figure 1. Color the cells in the UMAP plot by their graph-based cluster assignment
The 3D UMAP plot opens in a new data viewer session (Figure ?). Each point is a different cell and they are clustered together in the 3D plot based on how similar their expression profiles are across proteins and genes. Because a graph-based clustering task was performed upstream, a biomarker table is also displayed under the plot. This table lists the proteins and genes that are most highly expressed in each graph-based cluster. The graph-based clustering found 11 clusters, so there are 11 columns in the biomarker table.
Figure 1. Color the cells in the UMAP plot by their graph-based cluster assignment
The 3D UMAP plot opens in a new data viewer session (Figure ?). Each point is a different cell and they are clustered together in the 3D plot based on how similar their expression profiles are across proteins and genes. Because a graph-based clustering task was performed upstream, a biomarker table is also displayed under the plot. This table lists the proteins and genes that are most highly expressed in each graph-based cluster. The graph-based clustering found 11 clusters, so there are 11 columns in the biomarker table.
- Click and drag the 2D scatter plot icon from the Available plots card onto the canvas (Figure ?)
- Drop the 2D scatter plot to the right of the UMAP plot
 Figure 2. Add a 2D scatter plot and place it to the right of the UMAP plot
Figure 2. Add a 2D scatter plot and place it to the right of the UMAP plot
- Click Merged counts to use as data for the 2D scatter plot (Figure ?)
 Figure 3. Choose Merged counts data to draw the 2D scatter plot
A 2D scatter plot has been added to the right of the UMAP plot. The points in the 2D scatter plot are the same cells as in the UMAP, but they are positioned along the x- and y-axes according to their expression level for two protein markers: CD3_TotalSeqB and CD4_TotalSeqB, respectively (Figure ?).
Figure 3. Choose Merged counts data to draw the 2D scatter plot
A 2D scatter plot has been added to the right of the UMAP plot. The points in the 2D scatter plot are the same cells as in the UMAP, but they are positioned along the x- and y-axes according to their expression level for two protein markers: CD3_TotalSeqB and CD4_TotalSeqB, respectively (Figure ?).
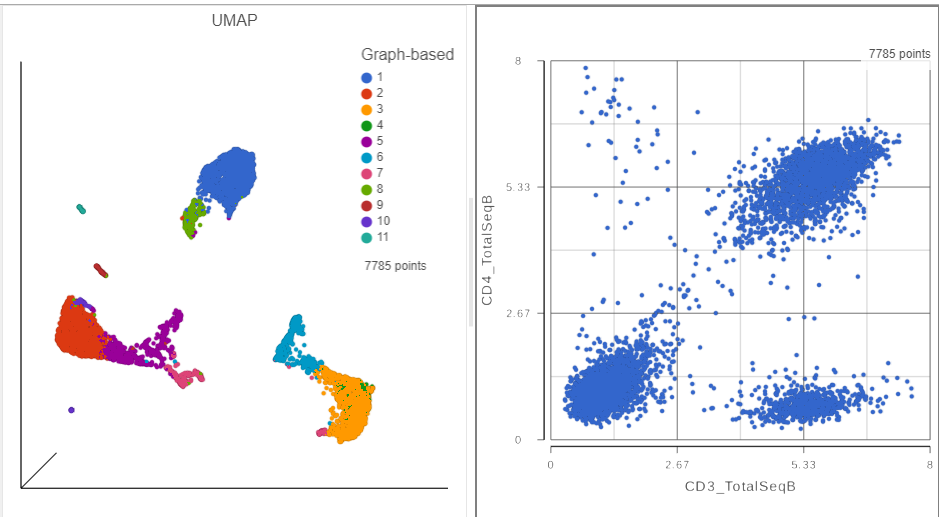 Figure 4. The canvas now has a 2D scatter plot next to the UMAP
Figure 4. The canvas now has a 2D scatter plot next to the UMAP
- In the Selection card on the right, click Rule to change the selection mode
- Click the blue circle next to the Add rule drop-down menu (Figure ?)
 Figure 5. Click the blue circle to change the data source for the rule selector
Figure 5. Click the blue circle to change the data source for the rule selector
- Click Merged counts to change the data source
- Choose CD3_TotalSeqB from the drop-down list (Figure ?)
 Figure 6. Choose the CD3_TotalSeqB protein marker as a selection rule
Figure 6. Choose the CD3_TotalSeqB protein marker as a selection rule
- Click and drag the slider on the CD3D_TotalSeqB selection rule to include the CD3 positive cells (Figure ?)
 Figure 7. Use the slider to select cells with positive expression for the CD3 protein marker
As you move the slider up and down, the corresponding points on both plots will dynamically update. The cells with a high expression for the CD3 protein marker (a marker for T cells) are highlighted and the deselected points are dimmed (Figure ?).
Figure 7. Use the slider to select cells with positive expression for the CD3 protein marker
As you move the slider up and down, the corresponding points on both plots will dynamically update. The cells with a high expression for the CD3 protein marker (a marker for T cells) are highlighted and the deselected points are dimmed (Figure ?).
 Figure 8. CD3+ cells are selected on both plots
Figure 8. CD3+ cells are selected on both plots
- Click Merged counts in the Data card on the left
- Click and drag CD8a_TotalSeqB onto the 2D scatter plot (Figure ?)
- Drop CD8_TotalSeqB onto the x-axis option
 Figure 9. Change the feature plotted on the x-axis to CD8_TotalSeqB
The CD3 positive cells are still selected, but now you can see how they separate into CD4 and CD8 positive populations (Figure ?).
Figure 9. Change the feature plotted on the x-axis to CD8_TotalSeqB
The CD3 positive cells are still selected, but now you can see how they separate into CD4 and CD8 positive populations (Figure ?).
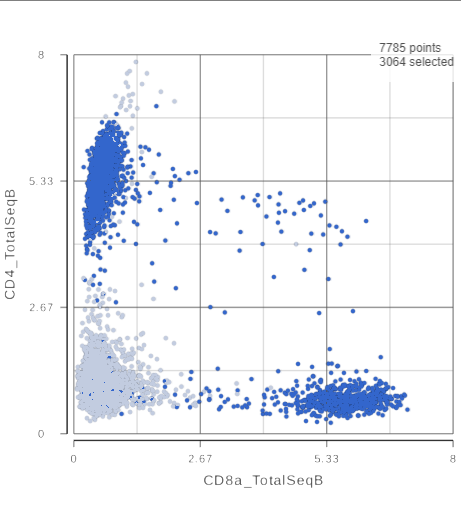 Figure 10. 2D scatter plot with CD4_TotalSeqB and CD8_TotalSeqB features on the axes
The simplest way to classifying cell types is to look for the expression of key marker genes or proteins. This approach is more effective with CITE-Seq data than with gene expression data alone as the protein expression data has a better dynamic range and is less sparse. Additionally, many cell types have expected cell surface marker profiles established using other technologies such as flow cytometry or CyTOF. Let's compare the resolution power of the CD4 and CD8A gene expression markers compared to their protein counterparts.
Figure 10. 2D scatter plot with CD4_TotalSeqB and CD8_TotalSeqB features on the axes
The simplest way to classifying cell types is to look for the expression of key marker genes or proteins. This approach is more effective with CITE-Seq data than with gene expression data alone as the protein expression data has a better dynamic range and is less sparse. Additionally, many cell types have expected cell surface marker profiles established using other technologies such as flow cytometry or CyTOF. Let's compare the resolution power of the CD4 and CD8A gene expression markers compared to their protein counterparts.
- Click the duplicate plot icon above the 2D scatter plot (Figure ?)
 Figure 11. Click the duplicate plot icon to make a copy of the 2D scatter plot
Figure 11. Click the duplicate plot icon to make a copy of the 2D scatter plot
- Click Merged counts in the Data card on the left
- Search for the CD4 gene
- Click and drag CD4 onto the duplicated 2D scatter plot
- Drop the CD4 gene onto the y-axis option
- Search for the CD8A gene
- Click and drag CD8A onto the duplicated 2D scatter plot
- Drop the CD8A gene onto the x-axis option
The second 2D scatter plot has the CD8A and CD4 mRNA markers on the x- and y-axis, respectively (Figure ?). The protein expression data has a better dynamic range than the gene expression data, making it easier to identify sub-populations.
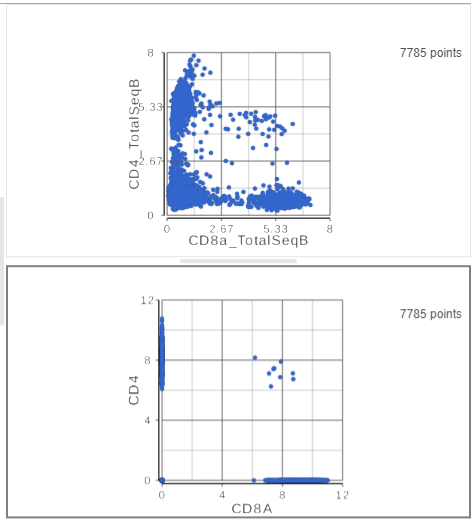 Figure 12. The second 2D scatter plot (bottom) has the CD8 and CD4 genes plotted against each other
Figure 12. The second 2D scatter plot (bottom) has the CD8 and CD4 genes plotted against each other
- On the first 2D scatter plot (with protein markers), click
 in the top right corner
in the top right corner - Manually select the cells with high expression of the CD4_TotalSeqB protein marker (Figure ?)
More than 2000 cells show positive expression for the CD4 cell surface protein.
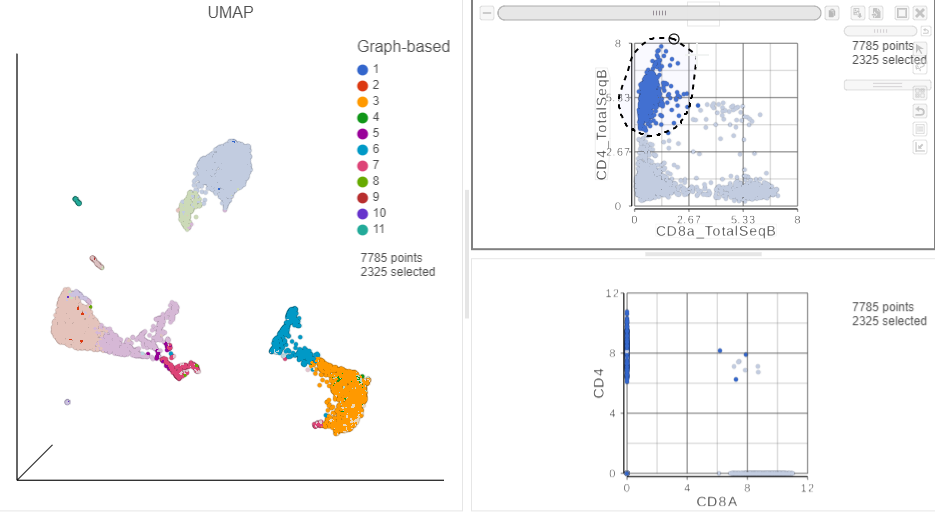 Figure 13. Draw a lasso to manually select CD4+ cells, based on protein expression
Figure 13. Draw a lasso to manually select CD4+ cells, based on protein expression
- Click
 in the top right of the plot to switch back to pointer mode
in the top right of the plot to switch back to pointer mode - Click on a blank spot on the plot to clear the selection
- On the second 2D scatter plot (with mRNA markers), click
 in the top right corner
in the top right corner - Manually select the cells with high expression of the CD4 gene marker (Figure ?)
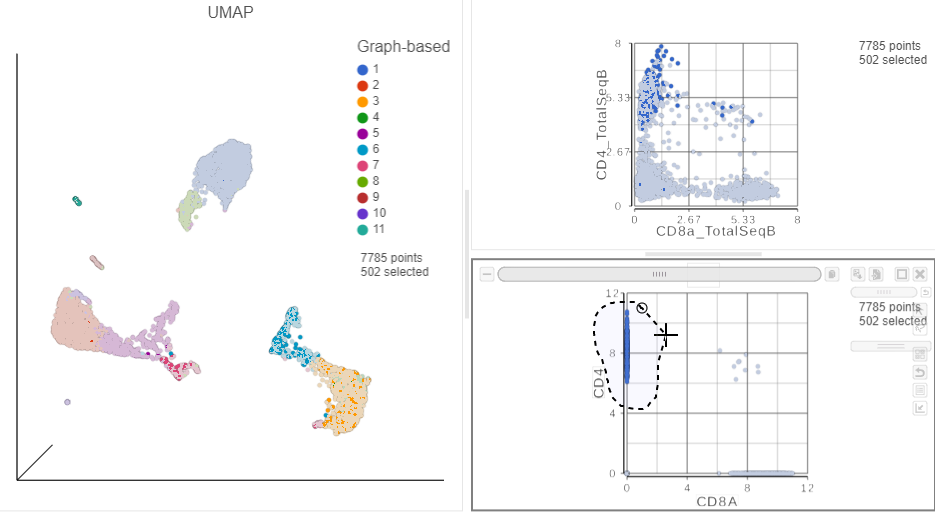 Figure 14. Draw a lasso to manually select CD4+ (mRNA) cells
This time, only 500 cells show positive expression for the CD4 marker gene. This means that the gene expression data is more sparse (i.e. there are more zeros), which further helps to reliably detect sub-populations.
Figure 14. Draw a lasso to manually select CD4+ (mRNA) cells
This time, only 500 cells show positive expression for the CD4 marker gene. This means that the gene expression data is more sparse (i.e. there are more zeros), which further helps to reliably detect sub-populations.
T cells
Based on the exploratory analysis above, most of the CD3 positive cells are in the group of cells in the bottom right corner of the UMAP plot. This is likely to be a group of T cells. We will now examine this group in more detail to identify T cell sub-populations.
- Click
 in the top right corner of both 2D scatter plots, to remove them from the canvas
in the top right corner of both 2D scatter plots, to remove them from the canvas - Click in the top right corner of the 3D UMAP plot
- Draw a lasso around the group of putative T cells (Figure ?)
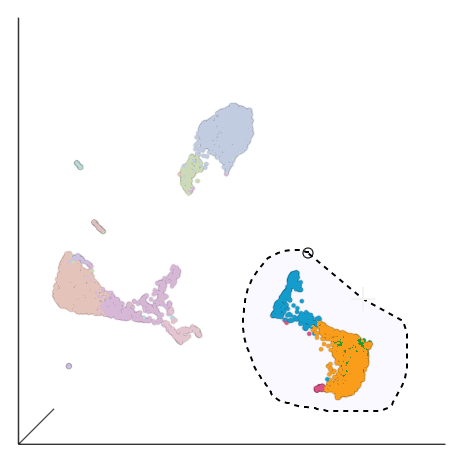 Figure 15. Select the group of putative T cells
Figure 15. Select the group of putative T cells
- Click
 to include the selected points
to include the selected points - Click
 in the top right of the plot to switch back to pointer mode
in the top right of the plot to switch back to pointer mode - Click and drag the plot to rotate it around
Deselected cells are excluded and the axes have been rescaled to give better resolution of the selected points (Figure ?). Note that the UMAP has not been recalculated, the axes have just been rescaled.
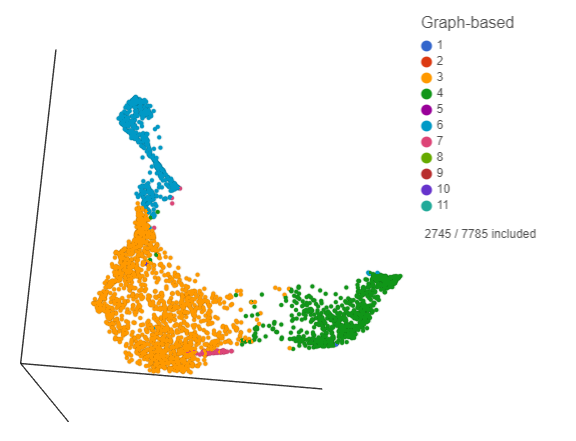 Figure 16. Group of putative T-cells
This group of putative T cells predominantly consists of cells assigned to graph-based clusters 3, 4, 6, and 7. Examining the biomarker table for these clusters can help us infer cell types.
Figure 16. Group of putative T-cells
This group of putative T cells predominantly consists of cells assigned to graph-based clusters 3, 4, 6, and 7. Examining the biomarker table for these clusters can help us infer cell types.
- Click and drag the bar between the UMAP plot and the biomarker table to resize the biomarker table to see more of it (Figure ?)
If you need to create more space on the canvas, hide the selection panel using the ![]() icon on the right and/or the
icon on the right and/or the ![]() icon to hide the menu on the left.
icon to hide the menu on the left.
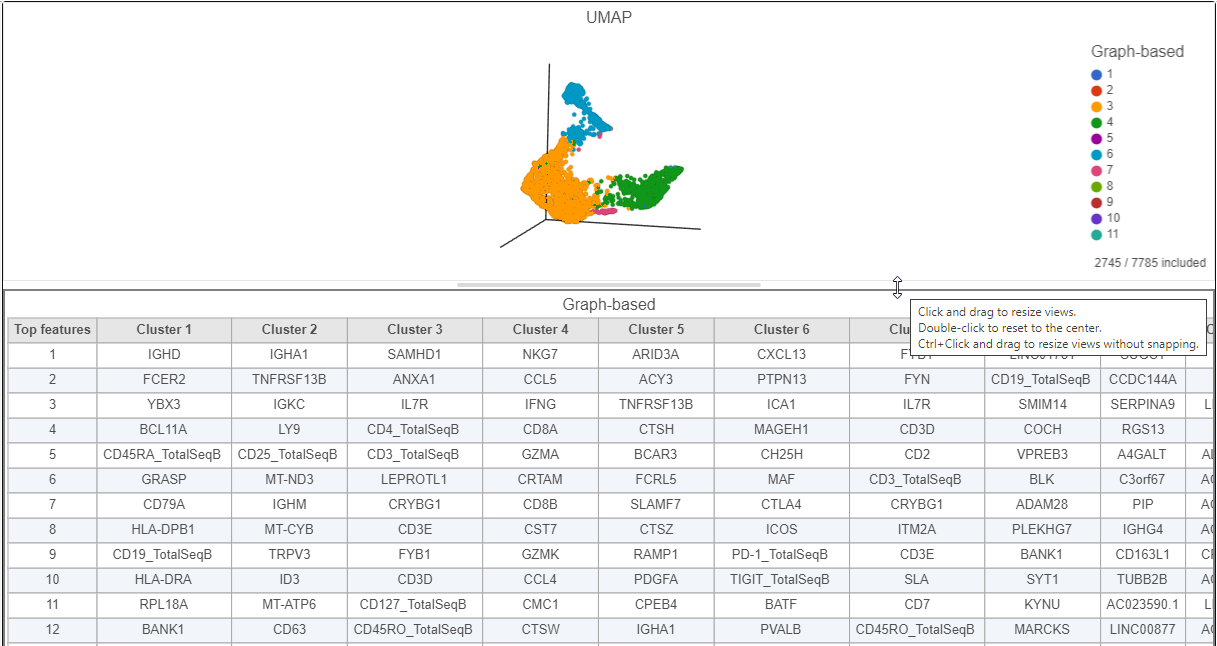 Figure 17. Resize plots to see more of the biomarker table
Figure 17. Resize plots to see more of the biomarker table
B cells
Additional Assistance
If you need additional assistance, please visit our support page to submit a help ticket or find phone numbers for regional support.


| Your Rating: |
|
Results: |

|
0 | rates |
Overview
Content Tools