Principal component analysis (PCA) can be performed to visualize clusters in the methylation data, but also serves as a quality control procedure; outliers within a group could suggest poor data quality, batch effects, mislabeled samples, or uninformative groupings.
- Select Plot PCA Scatter Plot from the QA/QC section of the Illumina BeadArray Methylation workflow to bring up a Scatter Plot tab
- Select 3. shRNA for Color by and 2. State for Size by
- Select (
 ) to enable Rotate Mode
) to enable Rotate Mode - Left click and drag to rotate the plot and view different angles (Figure 1)
Each dot of the plot is a single sample and represents the average methylation status across all CpG loci. We can see clear separation of naive and primed hPSCs from the naive hPSCs transduced with shRNA lentiviruses (shNANOG and shPOU5F1). One shNANOG sample might be an outlier, but we will not exclude it for this tutorial.
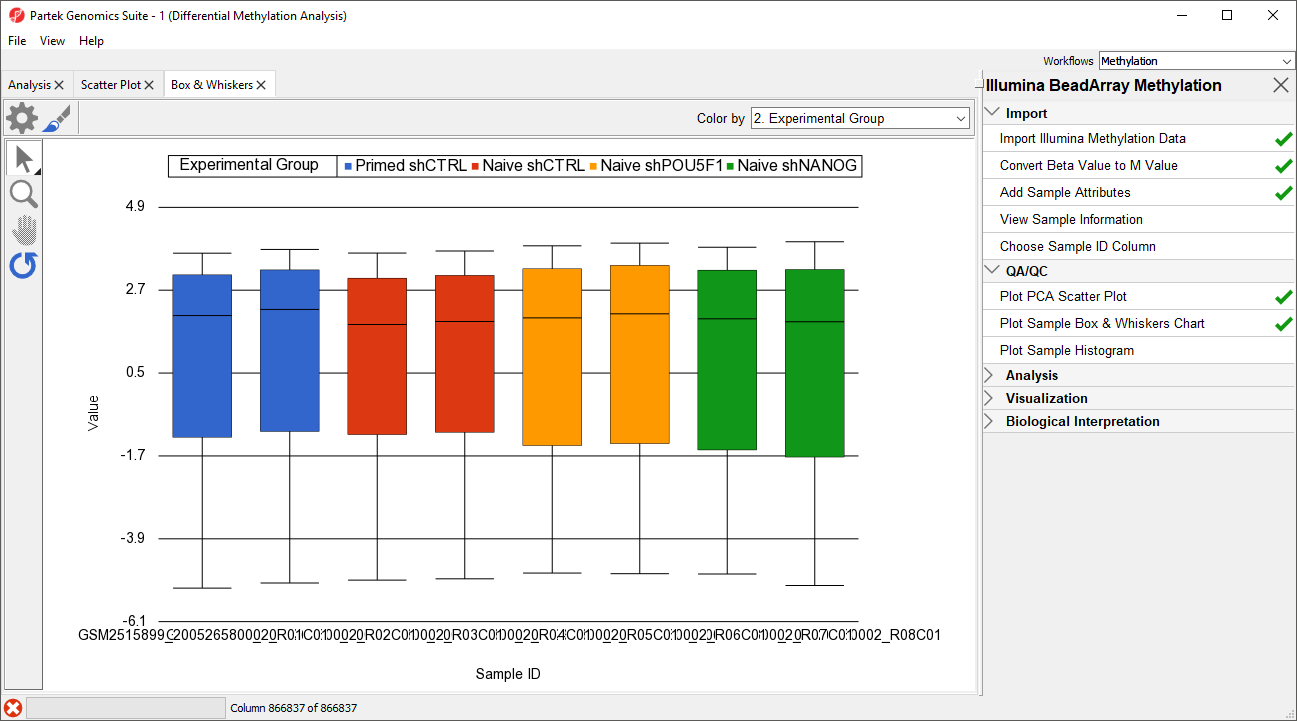
Figure 1. Principal components analysis (PCA) showing methylation profiles of the study samples. Each sample is represented by a dot, the axes are first three PCs, the number in parenthesis indicate the fraction of variance explained by each PC. The number at the top is the variance explained by the first three PCs. The samples are colored by levels of 3. shRNA and colored by 2. State
Next, distribution of M-values across the samples can also be inspected by a box-and-whiskers plot.
- Select Plot Sample Box and Whiskers Chart from the QA/QC section of the Illumina BeadArray Methylation workflow to bring up a Scatter Plot tab
Each box-and-whisker is a sample and the y-axis shows M-value ranges. Samples in this data set seem reasonably uniform (Figure 2).
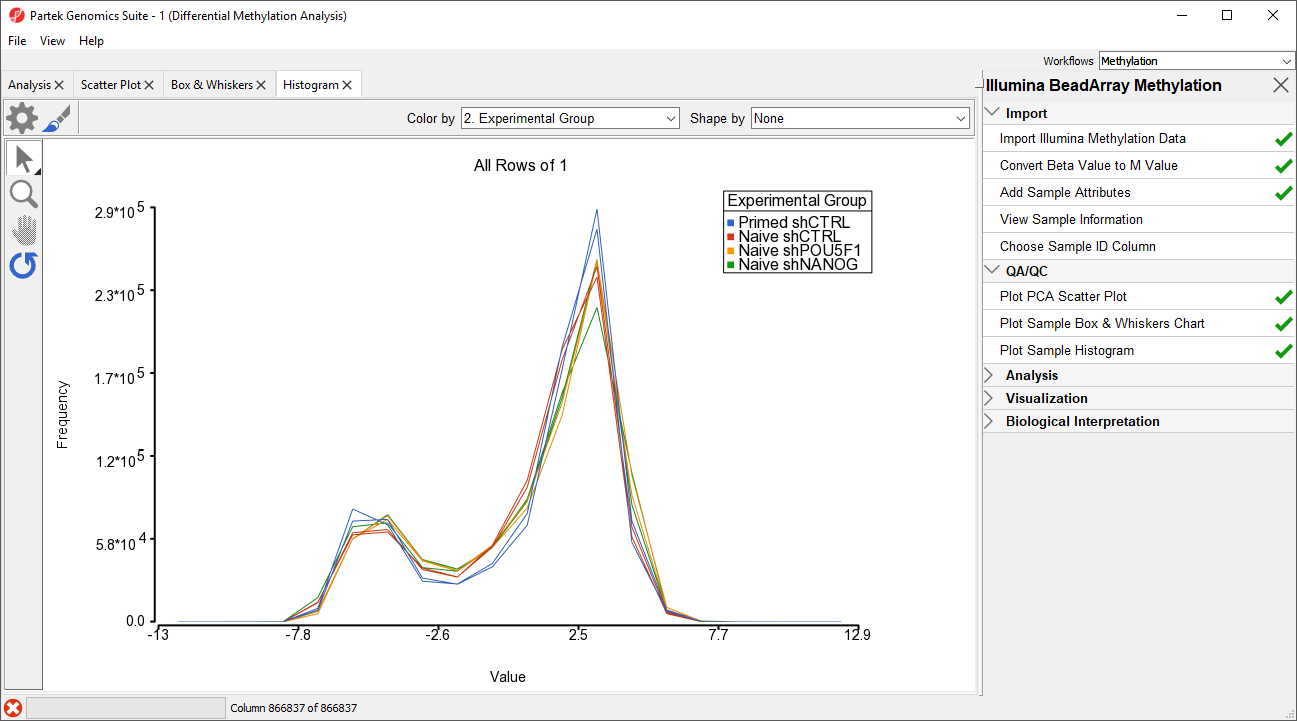
Figure 2. Box and whiskers plot showing distribution of M-values (y-axis) across the study samples (x-axis). Samples are colored by a categorical attribute (HPSC). The middle line is the median, box represents the upper and the lower quartile, while the whiskers correspond to the 90th and 10th percentile of the data
An alternative way to take a look at the distribution of M-values is a histogram.
- Select Plot Sample Histogram from the QA/QC section of the Illumina BeadArray Methylation workflow to bring up a Histogram tab
Again, no sample in the tutorial data set stands out (Figure 4).
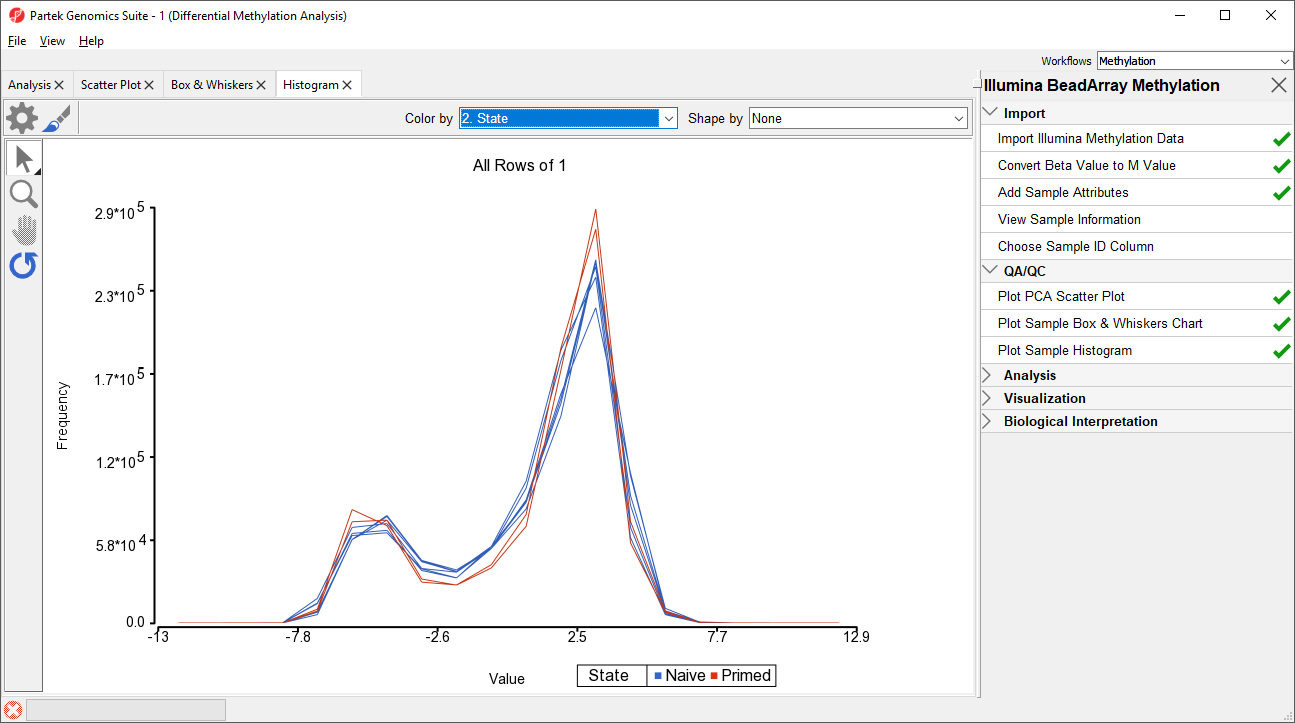 Figure 3. Sample histogram. Each sample is a line, M-values are on the horizontal axis and their frequencies on the vertical axis. Two peaks correspond to two probe types (I and II) present on the MethylationEPIC array. Sample colors correspond to a categorical attribute (i.e. State)
Figure 3. Sample histogram. Each sample is a line, M-values are on the horizontal axis and their frequencies on the vertical axis. Two peaks correspond to two probe types (I and II) present on the MethylationEPIC array. Sample colors correspond to a categorical attribute (i.e. State)
Additional Assistance
If you need additional assistance, please visit our support page to submit a help ticket or find phone numbers for regional support.


| Your Rating: |
|
Results: |

|
0 | rates |
Overview
Content Tools