Page History
...
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
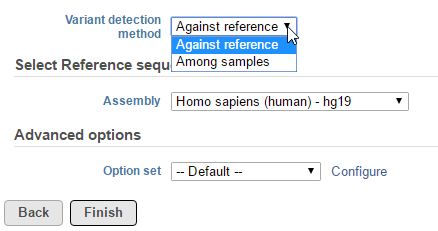
The detection method produce log-odds score, a high log-odds score for a reported SNV indicates a strong chance that the nucleotide is different from the reference sequence at that particular position in the detected sample. By default minimum log-odds on the reported SVN is 5. To change the value, click on Configure the Advanced options (Figure 2):
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
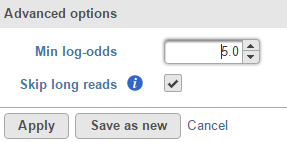
Variant detection among sample option is to compare base composition across all the samples in the input data node, but not to compare the reference sequence. A high log-odds score indicates a strong chance that at least one of the samples has a different base call at the position. This is useful in detection somatic mutations if there are 1 pair (two samples) in the input data.
Additional Assistance
| additional |
|---|
| -assistance |
|---|
|
Overview
Content Tools