Partek Genotype likelihood method utilizes observed base frequencies to calculate the genotype with the maximum likelihood at each genomic position based upon expected allele frequencies for heterozygotes and homozygotes and assuming a constant error probability. This method can identify single nucleotide variants but not insertions/deletions.
When a homozygous genotype would be observed, you would expect to observe nearly 100% of the homozygous allele. When the base frequencies of a heterozygous base are examined, you expected to observe nearly 50% for each allele. The observed base frequencies may deviate slightly from these numbers because call and alignment errors.
An example of expected allele probabilities using for an error probability of .01 is given below. If an error occurred (caused by base calling or mapping), assume each of the 4 alleles are equally likely to be observed with probability Perror (including alleles compatible with the genotype). Phom is the expected probability of observing the allele matching a homozygous genotype. Phet is the probability of observing each of the two alleles of a heterozygous genotype.
Perror = .01 / 4
Phom= 1.0 – 3 * Perror
Phet= .5 – Perror
The likelihood of a homozygous genotype AA given an observed base frequency F = {FA, Fc,FG,FT} can be expressed as:
L(AA | F, Perror) = PhomFA * Perror(FC + FG + FT)
The likelihood of a heterozygous genotype CT given an observed base frequency F can be expressed as:
L(CT | F, Perror) = Phet(FC + FT) * Perror(FA + FG)
The genotype, G, is assigned using maximum likelihood, and a log (base 10) odds ratio is calculated to aid in sorting.
Gmax = argsmax {L(G | F, Perrorr)}
Log Odds = log ( L(Gmax | F,Perrorr) / (1.0 – L(Gmax | F, Perror) )
If the Log Odds are undefined because of machine numeric representation limitations, then the log odds are capped at 106.
Partek Genotype Likelihood Dialog
Selecting Partek Genotype Likelihood from the context sensitive menu will bring up the task dialog, which contains three default sections: Variant detection method, Select Reference sequence, and Advanced options.
In the Variant detection method drop-down list, Against reference will compare base composition for each sample against the reference sequence assembly, independently (Figure 1).
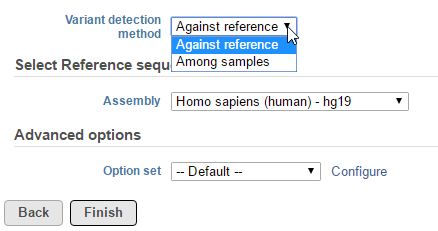 Figure 1. Selecting a variant detection method in the Partek Genotype Likelihood dialog
The reference sequence assembly should be selected from the drop-down list if the first input data file in the pipeline is a bam file. If the first input data file is raw sequence file, and the data is aligned in Partek Flow, then the reference sequence assembly used here is the same as the one used in the alignment step, there is no need to select the reference sequence.
Figure 1. Selecting a variant detection method in the Partek Genotype Likelihood dialog
The reference sequence assembly should be selected from the drop-down list if the first input data file in the pipeline is a bam file. If the first input data file is raw sequence file, and the data is aligned in Partek Flow, then the reference sequence assembly used here is the same as the one used in the alignment step, there is no need to select the reference sequence.
The detection method produce log-odds score, a high log-odds score for a reported SNV indicates a strong chance that the nucleotide is different from the reference sequence at that particular position in the detected sample. By default minimum log-odds on the reported SVN is 5. To change the value, click on Configure the Advanced options (Figure 2):
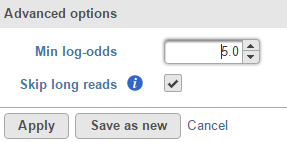 Figure 2. Advanced options in Partek genotype likelihood variant detection method
We recommend to check the Skip long reads option, which will ignore reads that span more than 0.1% of the reference.
Figure 2. Advanced options in Partek genotype likelihood variant detection method
We recommend to check the Skip long reads option, which will ignore reads that span more than 0.1% of the reference.
Variant detection among sample option is to compare base composition across all the samples in the input data node, but not to compare the reference sequence. A high log-odds score indicates a strong chance that at least one of the samples has a different base call at the position. This is useful in detection somatic mutations if there are 1 pair (two samples) in the input data.
Additional Assistance
If you need additional assistance, please visit our support page to submit a help ticket or find phone numbers for regional support.
Overview
Content Tools