Page History
...
If the alignment was generated in Partek® Flow®, the genome assembly will be displayed as text on the top of the page (Figure 1), you do not have the option to change the reference.
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
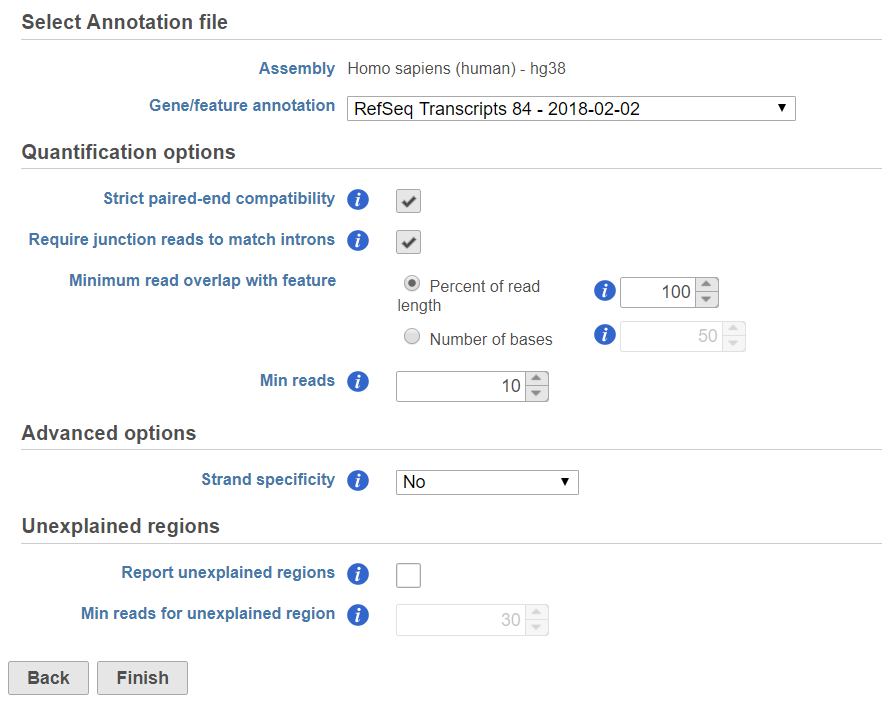
If the bam file is imported, you need to select the assembly with which the reads were aligned to, and which annotation model file you will use to quantify from the drop-down menus (Figure 2).
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
In the Quantification options section, when the Strict paired-end compatibility check button is selected, paired end reads will be considered compatible with a transcript only if both ends are compatible with the transcript. If it is not selected, reads with only one end have alignment that is compatible with the transcript will also be counted for the transcript .
...
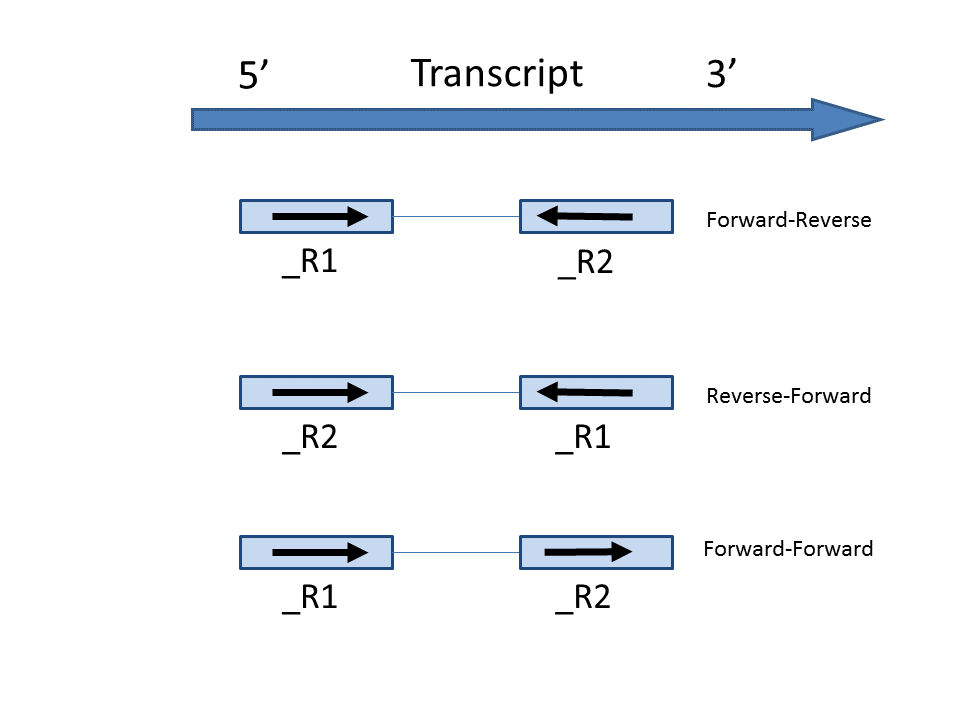
- Forward - Forward: Both ends of the read are matching the strand of the transcript. Generally colorspace data generated from SOLiD technology would follow this format
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
Minimum read overlap with feature can be specified in percentage of read length or number of bases. By default, a read has to be 100% within a feature. You can allow some overhanging bases outside the exonic region by modifying these parameters.
...
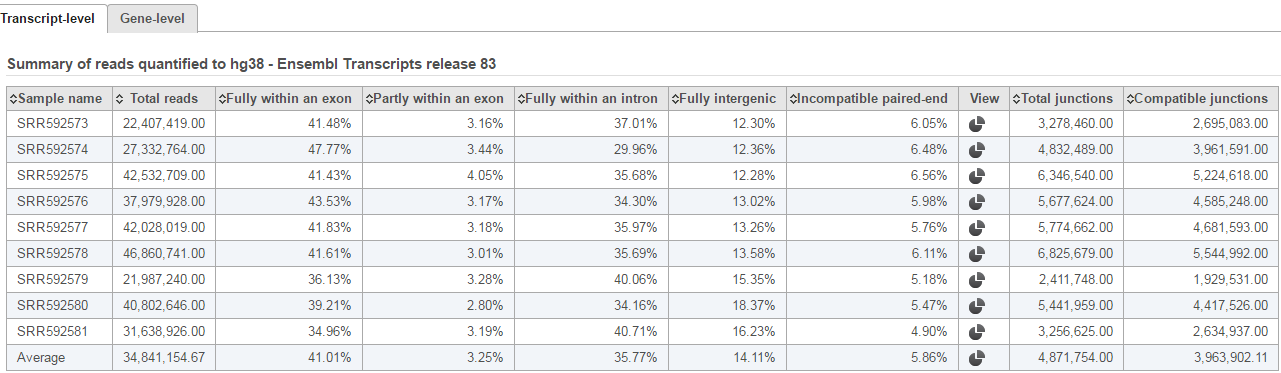
Depending on the annotation file, the output could be one or two data nodes. If the annotation file only contains one level of information, e.g. miRNA annotation file, you will only get one output data node. On the other hand, if the annotation file contains gene level and transcript level information, such as those from the Ensembl database, both gene and transcript level data nodes will be generated. If two nodes are generated, the Task report will also contain two tabs, reporting quantification results from each node. Each report has two tables. The first one is a summary table displaying the coverage information for each sample quantified against the specified transcriptome annotation (Figure 4).
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
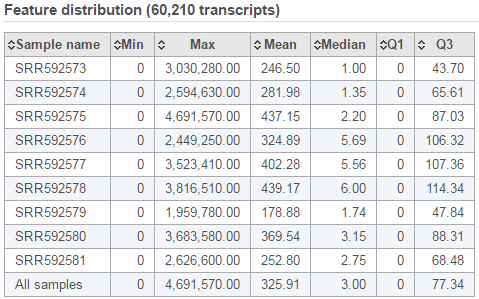
The second table contains feature distribution information on each sample and across all the samples, number of features in the annotation model is displayed on the table title (Figure 5).
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
The bar chart displaying the distribution of raw read counts is helpful in assessing the expression level distribution within each sample. The X-axis is the read count range, Y axis is the number of features within the range, each bar is a sample. Hovering your mouse over the bar displays the following information (Figure 6):
...
- Percentage of the features within the read count range
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|

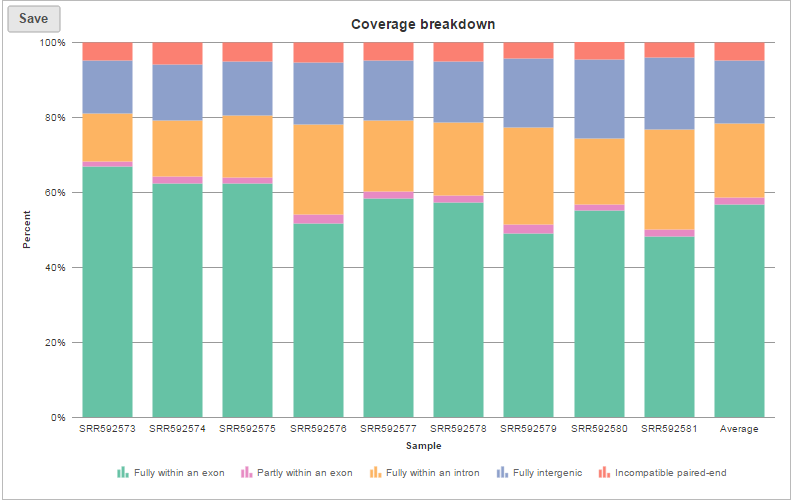
The coverage breakdown bar chart is a graphical representation of the reads summary table for each sample (Figure 7)
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
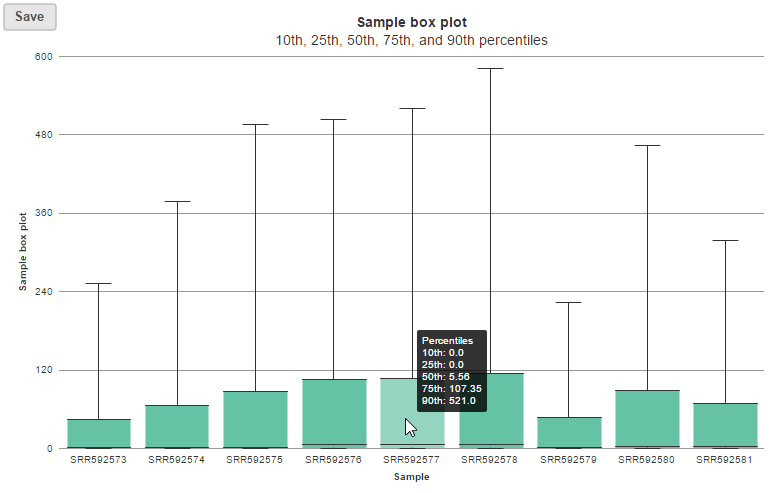
In the box-whisker plot, each box is a sample on X-axis, the box represents 25th and 75th percentile, the whiskers represent 10th and 90th percentile, Y-axis represents the read counts, when you hover over each box, detailed sample information is displayed (Figure 8).
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
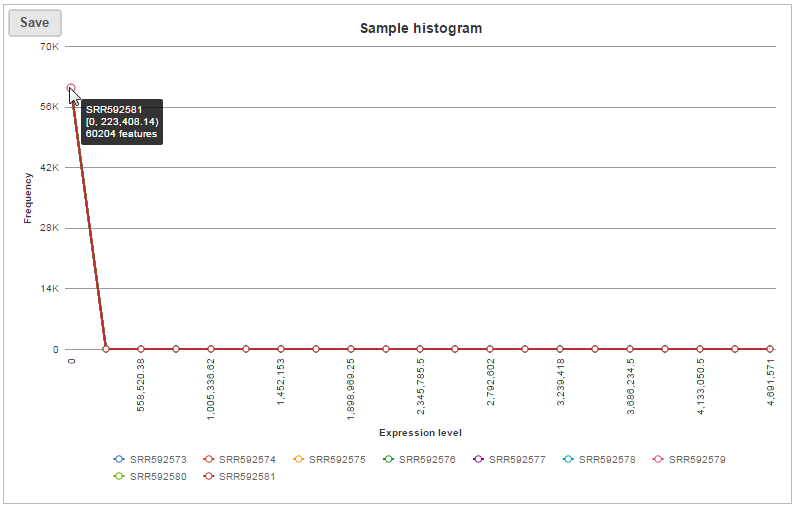
In sample histogram, each line represents a sample and the range of read counts are divided into 20 bins. Clicking on a sample in the legend will hide the line for that specific sample. Hovering over each circle displays detailed information about the sample and that specific bin (Figure 9). The information includes:
...
- Number of features within the read count range in the sample
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
The box whisker and sample histogram plots are helpful for understanding the expression level distribution across samples. This may indicate that normalization between samples might be needed prior to downstream analysis.
...
The Cufflinks task has three options that can be configured (Figure 10):
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
- Novel transcript: this option does not require any annotation reference, it will do de novo assembly to reconstruct transcripts and estimate their abundance
...
There are two parameters in Quantify to reference (Figure 11):
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
...
- Min coverage: will filter out any features (sequence names) that have fewer reads across all samples than the value specified
...
- Trapnell C, Williams B, Pertea G, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature Biotech. 2010; 28:511-515.
- Xing Y, Yu T, Wu YN, Roy M, Kim J, Lee C. An expectation-maximization algorithm for probabilistic reconstructions of full-length isoforms from splice graphs. Nucleic Acids Res. 2006; 34(10):3150-60.
| Additional assistance |
|---|
|
| Page Turner | ||
|---|---|---|
|
| Rate Macro | ||
|---|---|---|
|
Overview
Content Tools