Page History
...
Below is the notation that will be used to explain each method:
| Symbol | Meaning |
|---|---|
| S | Sample |
| F | FeatureFeature |
| Xsf | Value of sample S from feature F (if normalization is performed on a quantification data node, this would be the raw read counts) |
| TXsf | transformed value of Xsf |
| C | Constant value |
| b | Base of log |
- Absolute value
TXsf TXsf = | Xsf Xsf |
- Add
TXsf TXsf = Xsf Xsf + C
a constant value C needs to be specified
- Antilog
TXsf TXsf = bxsfbxsf
A log base value b needs to be specified from the drop-down list; any positive number can be specified when Custom value is chosen
- Divided by
When mean, median, Q1, Q3, std dev, or sum is selected, the corresponding statistics will be calculated based on the transform on sample or features option
Example: If transform on Samples is selected, Divide by mean is calculated as:
TXsf TXsf = XsfXsf/MsMs
where Ms is the mean of the sample.
Example: If transform on Features is selected, Divide by mean is calculated as:
TXsf TXsf = XsfXsf/MfMf
where Mf Mf is the mean of the feature.
- Log
TXsf = logbXsfTXsf = logbXsf
A log base value b needs to be specified from the drop-down list; any positive number can be specified when Custom value is chosen - Logit
TXsfTXsf=logblogb(XsfXsf/(1-XsfXsf))
A log base value b needs to be specified from the drop-down list; any positive number can be specified when Custom value is chosen
- Lower bound
A constant value C needs to be specified,
if Xsf Xsf is smaller than C, then TXsfTXsf= C; otherwise, TXsf TXsf = XsfXsf
- Multiply by
TXsf TXsf = Xsf Xsf x C
A constant value C needs to be specified - Quantile normalization, a rank based normalization method.
For instance, if transformation is performed on samples, it first ranks all the features in each sample. Say vector Vs Vs is the sorted feature values of sample S in ascending order, it calculates a vector that is the average of the sorted vectors across all samples --- VmVm, then the values in Vs Vs is replaced by the value in Vm Vm in the same rank. Detailed information can be found in [1]. - RPKM (Reads per kilobase of transcript per million mapped reads [2])
TXsf TXsf = (109 109 * XsfXsf)/(TMRsTMRs*LfLf)
Where Xsf Xsf is the raw read of sample S on feature F,
TMRs TMRs is the total mapped reads of sample S,
Lf Lf is the length of the feature F,
If quantification is performed on an aligned reads data node, total mapped reads is the aligned reads. If quantification is generated from imported read count text file, the total mapped reads is the sum of all feature reads in the sample.
If the feature is a transcript, transcript length Lf Lf is the sum of the lengths of all the exons. If the feature is a gene, gene length is the distance between the start position of the most downstream exon and the stop position of the most upstream exon. See Bullard et al. for additional comparisons with other normalization packages [3] - Subtract
When mean, median, Q1, Q3, std dev or sum is selected, the corresponding statistics will be calculated based on the transform on sample or features option
Example: If transform on Samples is selected, Subtract mean is calculated as:
TXsf TXsf = Xsf Xsf - MsMs
where Ms is the mean of the sample
Example: If transform on Features is selected, Subtract mean is calculated as:
TXsf TXsf = Xsf Xsf - MfMf
where Mf Mf is the mean of the feature
- TMM (Trimmed mean of M-values)
The scaling factors is produced according to the algorithm described in Robinson et al [4]. The paper by Dillies et al. [5] contains evidence that TMM has an edge over other normalization methods. - TPM (Transcripts per million as described in Wagner et al [6])
The following steps are performed:
- Normalize the reads by the length of feature, it generate reads per kilobase
RPKsf = Xsf / LfRPKsf = Xsf / Lf; - Sum up all the RPKsf in a sample
PRKs = ∑Ff=1 FRPKsfPRKs = ∑Ff=1 FRPKsf - Generate a scaling factor for each sample by normalizing the PRK of the sample to the sum PRK of all the samples
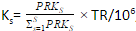 ,
,
where TR is the total reads across all samples - Divide raw reads by the scaling factor to get TPM
TXsf = Xsf/KsTXsf = Xsf/Ks
- Normalize the reads by the length of feature, it generate reads per kilobase
- Total count(Reads per million)
TXsf = (106 x Xsf106 x Xsf)/TMRsTMRs
where Xsf here is the raw read of sample S on feature F, and
TMRs is the total mapped reads of sample S.
If quantification is performed on an aligned reads data node, total mapped reads is the aligned reads. If quantification is generated from imported read count text file, the total mapped reads is the sum of all feature reads in the sample. - Upper quartile
The method is exactly the same as the LIMMA package [7].
The following is the simple summarization of the calculation:
...
Overview
Content Tools