Raw read counts are generated after quantification for each feature on all samples. These read counts need to be normalized prior to differential expression detection to ensure that samples are comparable.
This chapter covers the implementation of each normalization method. The Normalize counts option is available on the context-sensitive menu (Figure 1) upon selection of any quantified output data node or an imported count matrix:
- Gene counts
- Transcript counts
- MicroRNA counts
- Cufflinks quantification
- Quantification

Figure 1. When a data node containing quantified data is selected, Normalization becomes available on the context sensitive menu
The format of the output is the same as the input data format, the node is called Normalized counts. This data node can be selected and normalized further using the same task.
Selecting Methods
Select whether you want your data normalized on a per sample or per feature basis (Figure 2). Some transformations are performed on each value independently of others e.g. log transformation, and you will get an identical result regardless of your choice.
Figure 2. Transformation can be done on samples or on features
The following normalization methods will generate different results depending on whether the transformation was performed on samples or on features:
- Divided by mean, median, Q1, Q3, std dev, sum
- Subtract mean, median, Q1, Q3, std dev, sum
- Quantile normalization
Note that each task can only perform normalization on samples or features. If you wish to perform both transformations, run two normalization tasks successively. To normalize the data, click on a method from the left panel, then drag and drop the method to the right panel. Add all normalization methods you wish to perform. Alternatively, you can click on the green plus button (![]() ) on each method to add it. Multiple methods can be added to the right panel and they will be processed in the order they are listed. You can change the order of methods by dragging each method up or down. To remove a method from the Normalization order panel, click the minus button (
) on each method to add it. Multiple methods can be added to the right panel and they will be processed in the order they are listed. You can change the order of methods by dragging each method up or down. To remove a method from the Normalization order panel, click the minus button (![]() ) to the right of the method. Click Finish, when you are done choosing the normalization methods you have chosen.
) to the right of the method. Click Finish, when you are done choosing the normalization methods you have chosen.
Recommended Methods
For some data nodes, recommended methods are available:
- Data nodes resulting from Quantify to annotation model (Partek E/M) or Quantify to reference (Partek E/M) are raw read counts, the recommendation is Total Count, Add 0.0001
- Cufflinks quantification data node output FPKM normalized read counts, the recommendation is Add 0.0001
If available, the Recommended button will appear. Clicking the button will populate the right panel (Figure 3).
Figure 3. Normalization using Partek’s recommended method
Normalization Methods
Below is the notation that will be used to explain each method:
| Symbol | Meaning |
|---|---|
| S | Sample (or cell for single cell data node) |
| F | Feature |
| Xsf | Value of sample S from feature F (if normalization is performed on a quantification data node, this would be the raw read counts) |
| TXsf | transformed value of Xsf |
| C | Constant value |
| b | Base of log |
- Absolute value
TXsf = | Xsf | - Add
TXsf = Xsf + C
a constant value C needs to be specified - Antilog
TXsf = bxsf
A log base value b needs to be specified from the drop-down list; any positive number can be specified when Custom value is chosen - Arcsinh
TXsf =arcsinh (Xsf)
The hyperbolic arcsine (arcsinh) transformation is often used on flow cytometry data - CLR (centered log ratio)
TXsf =ln((Xsf +1)/geom (Xsf +1) +1)
geom is geometric mean of either observation or feature. This method can be applied on protein expression data. - CPM (counts per million)
TXsf = (106 x Xsf)/TMRs
where Xsf here is the raw read of sample S on feature F, and TMRs is the total mapped reads of sample S.
If quantification is performed on an aligned reads data node, total mapped reads is the aligned reads. If quantification is generated from imported read count text file, the total mapped reads is the sum of all feature reads in the sample. - Divided by
When mean, median, Q1, Q3, std dev, or sum is selected, the corresponding statistics will be calculated based on the transform on sample or features option
Example: If transform on Samples is selected, Divide by mean is calculated as:
TXsf = Xsf/Ms
where Ms is the mean of the sample.
Example: If transform on Features is selected, Divide by mean is calculated as:
TXsf = Xsf/Mf
where Mf is the mean of the feature. - Log
TXsf = logbXsf
A log base value b needs to be specified from the drop-down list; any positive number can be specified when Custom value is chosen - Logit
TXsf=logb(Xsf/(1-Xsf))
A log base value b needs to be specified from the drop-down list; any positive number can be specified when Custom value is chosen - Lower bound
A constant value C needs to be specified,
if Xsf is smaller than C, then TXsf= C; otherwise, TXsf = Xsf - Median ratio (DESeq2 only), Median ratio (edgeR)
These approaches are slightly different implementations of the method proposed by Anders and Huber (2010). The idea is as follows: for each feature, its expression is divided by the feature geometric mean expression across the samples. Then, for a given sample, one takes median of these ratios across the features and obtains a sample specific size factor. The normalized expression is equal to the raw expression divided by the size factor.
Median ratio (DESeq2 only) is present in R, DESeq2 package, under the name of "ratio". This method should be selected if DESeq2 differential analysis will be used for downstream analysis, since it is not per million scale, not recommended to be used in any other differential analysis methods except for DESeq2.
Median ratio (edgeR) is present in R, edgeR package under the name of “RLE”. It is very similar to Median ratio (DESeq2 only) method, but it uses per million scale. - Multiply by
TXsf = Xsf x C
A constant value C needs to be specified - Poscounts (Deseq2 only)
Deseq2 size factor estimate option. Comparing with Median ratio, poscount method can be used when all genes contain a sample with a zero. It calculates a modified geometric mean by taking the nth root of the product of the non-zero counts. It is not per million scale. Here is the details. - Quantile normalization, a rank based normalization method.
For instance, if transformation is performed on samples, it first ranks all the features in each sample. Say vector Vs is the sorted feature values of sample S in ascending order, it calculates a vector that is the average of the sorted vectors across all samples --- Vm, then the values in Vs is replaced by the value in Vm in the same rank. Detailed information can be found in [1]. - Rank
This transformation replaces each value with its rank in the list of sorted values. The smallest value is replaced by 1 and the largest value is replaced by the total number of non-missing values, N. If there are no tied values, the results in a perfectly uniform distribution. In the case of ties, all tied values receive the mean rank. - Rlog
Regularied log transformation is the method implemented in DESeq2 package under the name of rlog. It applies a transformation to remove the dependence of the variance on mean. It should not be applied on zero inflated data such as single cell RNA-seq raw count data. The output of this task should not be used for differential expression analysis, but rather for data exploration, like clustering etc. - Round
Round the value to the nearest integer.
- RPKM (Reads per kilobase of transcript per million mapped reads [2])
TXsf = (109 * Xsf)/(TMRs*Lf)
Where Xsf is the raw read of sample S on feature F,
TMRs is the total mapped reads of sample S,
Lf is the length of the feature F,
If quantification is performed on an aligned reads data node, total mapped reads is the aligned reads. If quantification is generated from imported read count text file, the total mapped reads is the sum of all feature reads in the sample.
If the feature is a transcript, transcript length Lf is the sum of the lengths of all the exons. If the feature is a gene, gene length is the distance between the start position of the most downstream exon and the stop position of the most upstream exon. See Bullard et al. for additional comparisons with other normalization packages [3]
For paired reads, the normalization option will show up as FPKM (Fragments per kilobase per million mapped reads) rather than RPKM. However, the calculations are the same. - Subtract
When mean, median, Q1, Q3, std dev or sum is selected, the corresponding statistics will be calculated based on the transform on sample or features option
Example: If transform on Samples is selected, Subtract mean is calculated as:
TXsf = Xsf - Ms
where Ms is the mean of the sample
Example: If transform on Features is selected, Subtract mean is calculated as:
TXsf = Xsf - Mf
where Mf is the mean of the feature - TMM (Trimmed mean of M-values)
The scaling factors is produced according to the algorithm described in Robinson et al [4]. The paper by Dillies et al. [5] contains evidence that TMM has an edge over other normalization methods. The reference sample is randomly selected. When perform the trimming, for M values (fold change), the upper 30% and lower 30% are removed; for A values (absolute expression), the upper 5% and lower 5% are removed. - TPM (Transcripts per million as described in Wagner et al [6])
The following steps are performed:
- Normalize the reads by the feature length. Here length is measured in kilobases but the final TPM values do not depend on the length unit.
RPKsf = Xsf / Lf; - Obtain a scaling factor for sample s as
Ks = 10-6 ∑Ff=1 RPKsf - Divide raw reads by the length and the scaling factor to get TPM
TXsf = Xsf / Lf / Ks
- Normalize the reads by the feature length. Here length is measured in kilobases but the final TPM values do not depend on the length unit.
- Upper quartile
- The method is exactly the same as the LIMMA package [7].
The following is the simple summarization of the calculation:
- Remove all the features that have 0 reads in all samples.
- Calculate the effective library size per sample: effective library size = (raw library size (in millions))*((upper quartile for a particular sample)/ (geometric mean of upper quartiles in all the samples))
- Get the normalized counts by dividing the raw counts per feature by the effective library size (for the respective sample)
Normalization Report
The Normalization report includes the Normalization methods used, a Feature distribution table, Box-whisker plots of the Expression signal before and after normalization, and Sample histogram charts before and after normalization. Note that all visualizations are disabled for results with more than 30 samples.
Normalization methods
A summary of the normalization methods performed. They are listed by the order they were performed.
Feature distribution table
A table that presents descriptive statistics on each sample, the last row is the grand statistics across all samples (Figure 4).
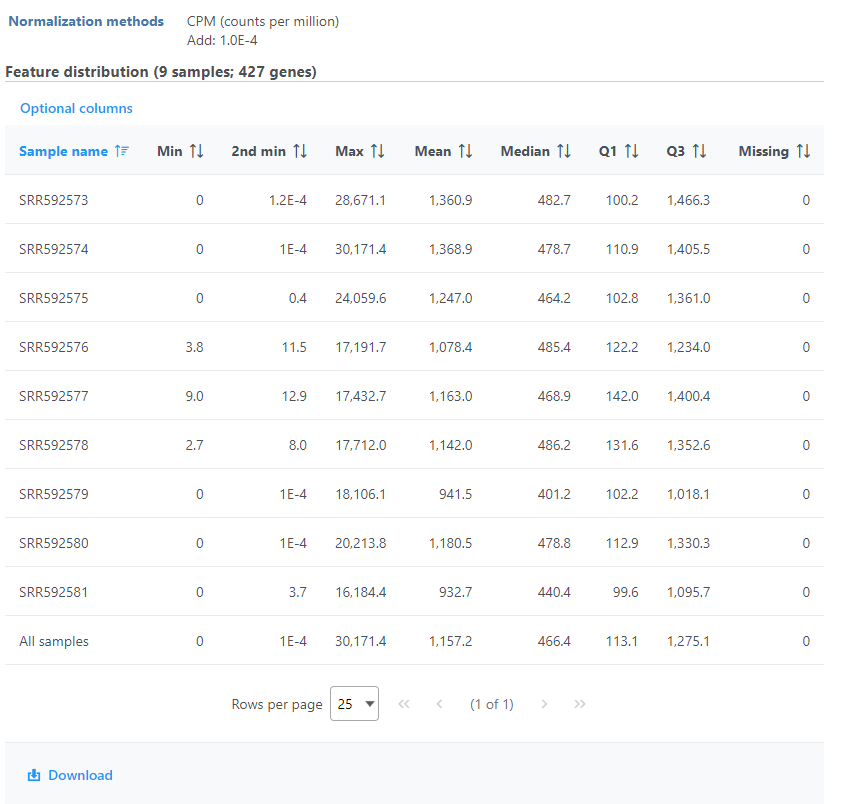
Figure 4. Feature distribution statistic information on each sample and across all the samples
Expression signal
These box-whisker plots show the expression signal distribution for each sample before and after normalization. When you mouse over on each bar in the plot, a balloon would show detailed percentile information (Figure 5).
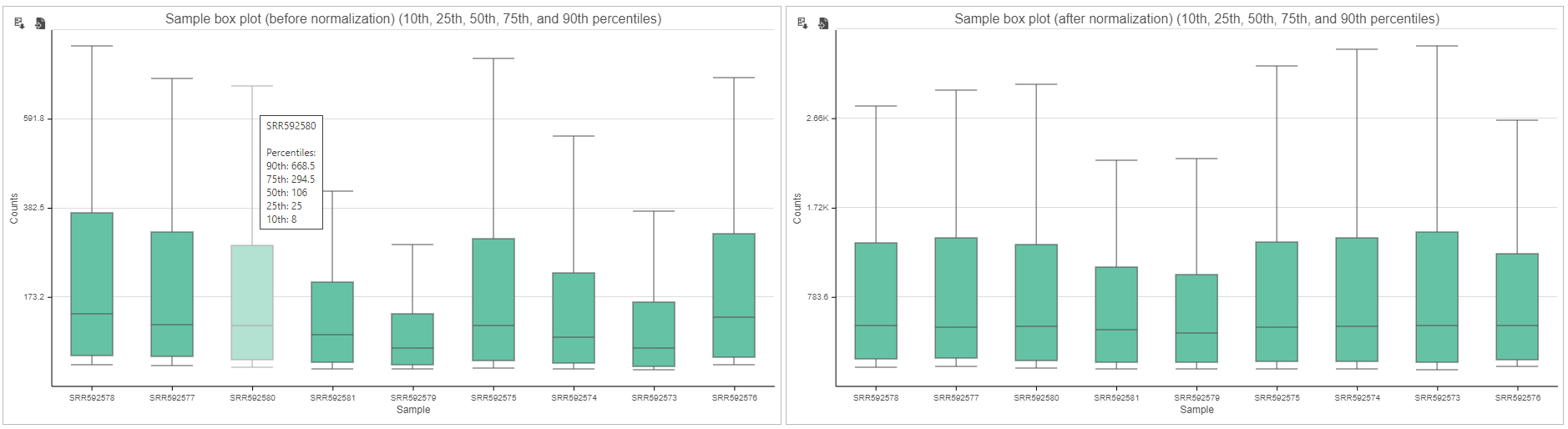
Figure 5. Box-whisker plot displays expression signal distribution for each sample
Sample histogram
A histogram is displayed for data before and after it is normalized. Each line is a sample, where the X axis is the range of the data in the node and the Y-axis is the frequency of the value within the range. When you mouse over a circle which represent a center of an interval, detailed information will appear in a balloon (Figure 6). It includes:
- The sample name.
- The range of the interval, “[ “represent inclusive, “)” represent exclusive.
- The frequency value within the interval
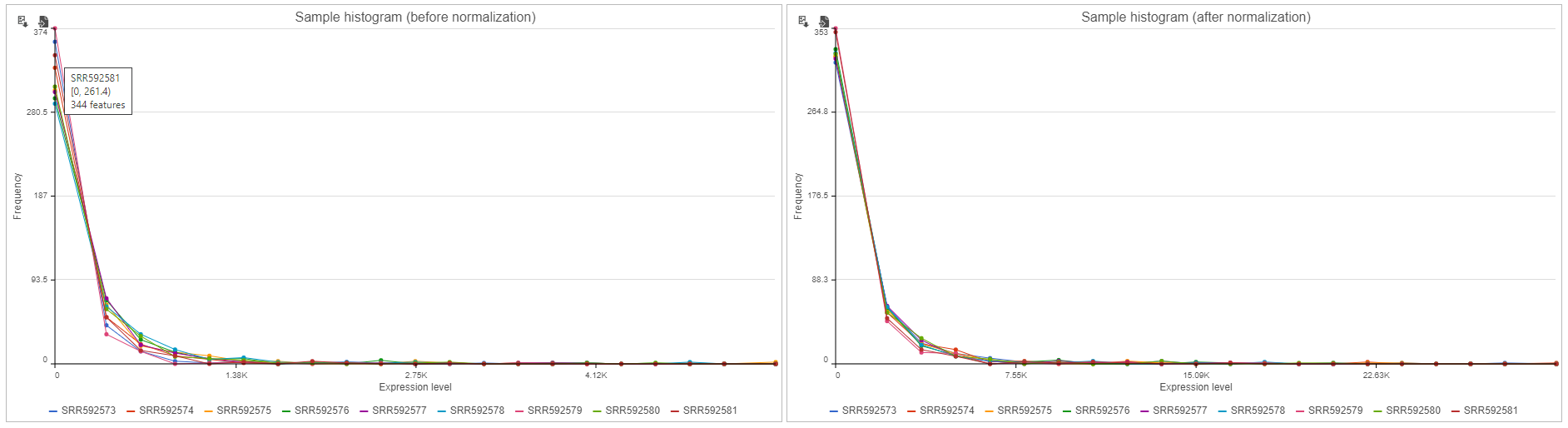
Figure 6. Sample histogram. Mousing over shows detailed information about the interval. This includes sample name, range and frequency of the selected sample.
References
- Bolstad BM, Irizarry RA, Astrand M, Speed, TP. A Comparison of Normalization Methods for High Density Oligonucleotide Array Data Based on Bias and Variance. Bioinformatics. 2003; 19(2): 185-193.
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008; 5(7): 621–628.
- Bullard JH, Purdom E, Hansen KD, Dudoit S. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinformatics. 2010; 11: 94.
- Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010; 11: R25.
- Dillies MA, Rau A, Aubert J et al. A comprehensive evaluation of normalization methods for Illumina high-throughput RNA sequencing data analysis. Brief Bioinform. 2013; 14(6): 671-83.
- Wagner GP, Kin K, Lynch VJ. Measurement of mRNA abundance using RNA-seq data. Theory Biosci. 2012; 131(4): 281-5.
- Ritchie ME, Phipson B, Wu D et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015; 43(15):e97.
Additional Assistance
If you need additional assistance, please visit our support page to submit a help ticket or find phone numbers for regional support.


| Your Rating: |
|
Results: |

|
41 | rates |
Overview
Content Tools
1 Comment
Melissa del Rosario
author: wxw