...
| Numbered figure captions |
|---|
| SubtitleText | Quantify to annotation model(Partek E/M) dialog |
|---|
| AnchorName | em |
|---|
|
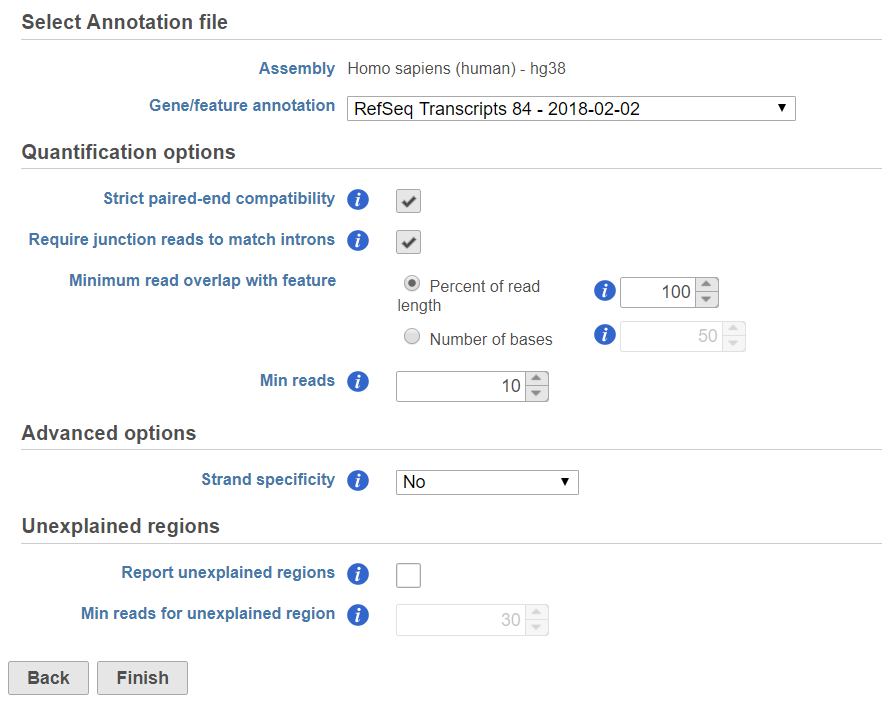 Image Removed Image Removed Image Added Image Added
|
If the bam file is imported, you need to select the assembly with which the reads were aligned to, and which annotation model file you will use to quantify from the drop-down menus (Figure 2).
...
| Numbered figure captions |
|---|
| SubtitleText | Specify the genome assembly with which the bam files are generated from and transcriptome annotation from the drop-down menu |
|---|
| AnchorName | specify-annot |
|---|
|
Image Modified |
In the Quantification options section, when the Strict paired-end compatibility check button is selected, paired end reads will be considered compatible with a transcript only if both ends are compatible with the transcript. If it is not selected, reads with only one end have alignment that is compatible with the transcript will also be counted for the transcript ....