Page History
| Table of Contents | ||||||
|---|---|---|---|---|---|---|
|
Motif detection tasks identify transcription factor binding identifies enriched sequence motifs in peak regions generated by ChIP-Seq and ATAC-Seq data. Partek Flow includes tasks that allow Search for known motifs, which allows users to search for known motifs from a database as well as to detect user-specified set or a database, and Detect de novo motifs, which can identify novel motifs. These tasks can be invoked on data nodes with genomic regions as features (not genes or transcripts).
Search for
...
known motifs
Given a set of genomic regions, sequence motif can be search Search for known motifs can search for enrichment based on a string sequence provided by user or alignment matrix from using a sequence database like JASPAR.
- Click on a
...
- Peaks data node
...
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
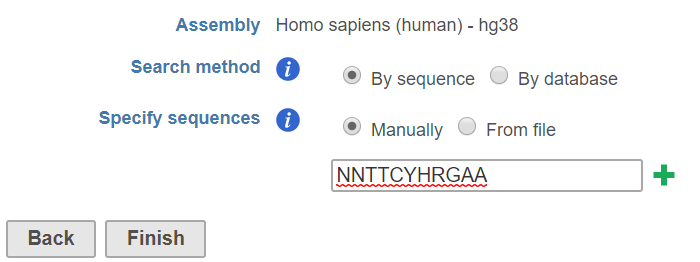
When choose the search method by sequence, there are two options:
...
- Click Motif detection in the toolbox
- Click Search for known motifs
The configuration dialog offers two search methods, By sequence and By database.
By sequence
By sequence has two options (Figure 1):
Manually: the user can manually specify the sequences, click on green plus button to add multiple sequences to search.. Multiple sequences can be added by clicking the ![]() button
button
From file: use can specify a text file (.txt) that contains a list of sequences, one row per sequence.
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
The By sequence option uses a string search tool will to return all positions in the set of genomic regions that match the given stringsstring(s). The string match is case insensitive, meaning if you search for ATCG, you may get atcG as a match. Nucleotides that are lower case have been "repeat masked", meaning they are located in a repetitive region of the genome. Your search string may contain any of the characters from the IUPAC nucleotide code . For example, if you search for WAAA, you may get back AAAA or TAAA (or any variantion variation of upper and lower cases), since because W represents A or T.
By database
The By database option uses an alignment matrix to match sequences against a motif database. We distribute the JASPAR database, but you can add any custom or public motif database.
Matching is determined using an alignment matrix. Alignment matrices are often used in literature to model transcription factor binding sites, alignment matrices are matrices of nucleotide counts per position [1]. Each instance of the motif is aligned to each other and the number of nucleotides at each position is counted and summarized in an alignment matrix. All positions from the set of genomic regions are scored against the alignment matrix. The score represents how likely the position is an an instance of the motif. A quality cutoff is used to determine which sequences in the regions are instances of the motif. The scoring scheme and quality cutoff are similar to [2] and it briefly described below:
Let M be a motif of length L consisting of N motif instances.Let A be a 4XL alignment matrix such that ai,j is the count of letter i at position j. Let Bi be the background frequency of letter i (calculated as the number of nucleotides i in the regions divided by the total oligonucleotides in the regions). Let S be a sequence of length L. The score of S given the alignment matrix is
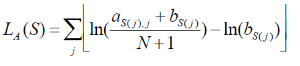
Equation 1
Let h be the maximum of LA. The quality score of a sequence is calculated as QA(S) =LA(S)/h. A quality score of 1 corresponds to a sequence with the most likely base at each position of the alignment matrix. User The user will specify a threshold QA. all All sequences that have a score TA>QA *h will be reported.The report
Task report
The Search for known motifs task report contains summary and detail tabs:.
| Numbered figure captions | |
|---|---|
|
...
|
...
| |||
|
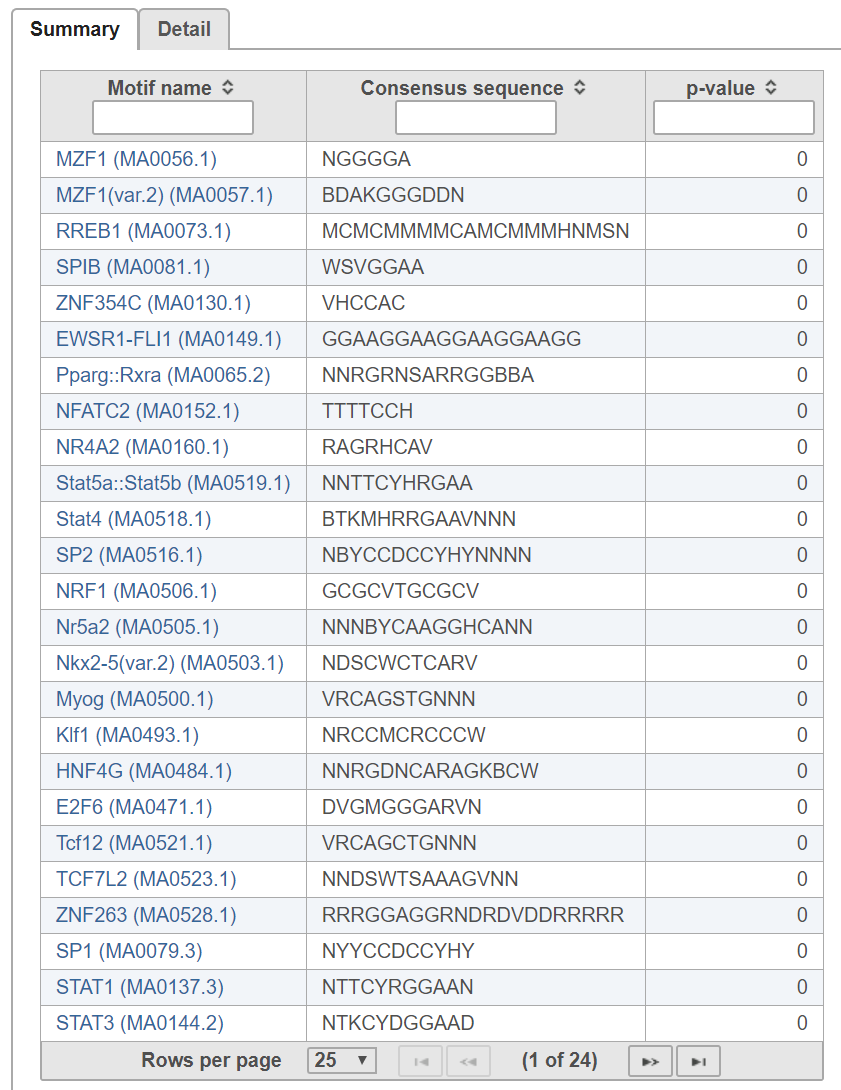
In the summary table tab (Figure 2), each row is a motif in the search database specified. Click Clicking on the motif name will take to opens the JASPAR database page to view more with detailed information about the motif.
The p-value indicates whether instances of the motif are enriched in the input regions.
The p-value is calculated as follows:
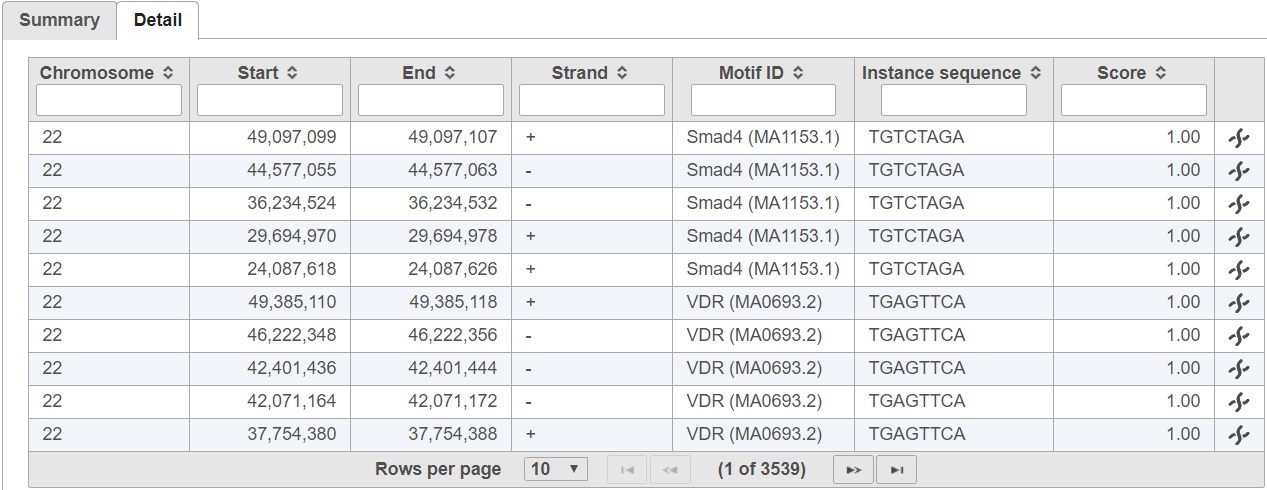
The probability PExpected of a sequence having a score above TA is calculated under the assumption that the base are i.i.d. according to the background distribution B. Let NTrials be the number of sequences compared to the alignment matrix. The expected number of occurrences of the motif in the regions is PExpected * NTrials. The p-value of observing NActual instances with a score above TA is calculated based on the binomial distribution, where NTrials is the number of trials and PExpected is the probability of success. A low p-value indicates that the regions are enriched with instances of the motif.In the detail table, each row is a location containing a motif sequence with quality score
The detail tab lists motif sequence locations on rows and includes the quality score for each instance (Figure 3).
| Numbered figure captions | |
|---|---|
|
...
| |||
|
...
Detect de novo motifs
Detect de novo motifs can be used to identify novel motifs that are enriched in the input regions.
- Click on
...
- a Peaks data node
- Click Motif detection in the toolbox
- Click Detect de novo motifs
...
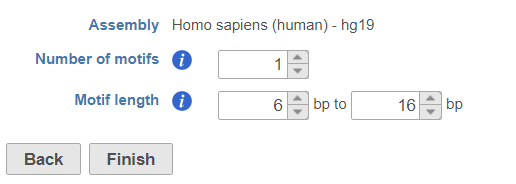
- Specify the number of motifs to report (Figure 4)
...
- Specify the
...
- range of
...
- motif length to search
| Numbered figure captions | ||
|---|---|---|
|
...
| |||
|
Motif discovery is done using uses Gibbs motif sampling. Partek Flow's The implementation of the Gibbs motif sampling in Partek Flow is based on Neuwal, et all [3]. the The Gibbs sampling method is a stochastic procedure that attempts to find the subset of sequences within the regions that maximizes the log likelihood ratio (LLR)
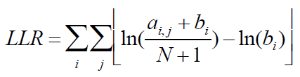
Equation 2
This is done by repeating the below following two steps until convergencesconvergence:
A. Given the alignment matrix from step B, search for location in the set of regions that score highly compared to the alignment matrix using Equation 1
...
The Gibbs sampler is run on a range of the motif sizes specified by the user. The motif with the greatest average LLR (LLR /length) is returned. To find N motifs ina in a set of regions, the Gibbs sampling method is run N times. The motif instances found from the previous run of the Gibbs sampler are removed before performing the next run.
Task report
The task report contains includes summary and detail tabs:.
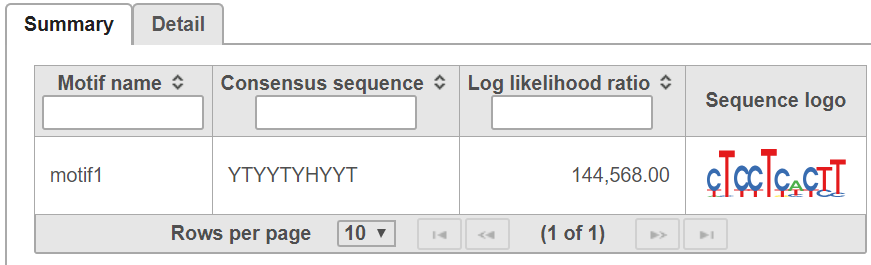
In summary table, it contains motif The summary tab gives the consensus sequence and sequence logo for each detected motif (Figure 5):.
| Numbered figure captions | |
|---|---|
|
...
| |||
|
The detail table contains the locations of tab is similar to the detail tab in Search for known motifs and lists the location of each motif sequence (Figure 6):.
| Numbered figure captions | |
|---|---|
|
...
| |||
|

...
References
Reference
- Hertz, GZ., & Stormo, G.D. Identifying DNA and protein patterns with statistically significant alignments of multiple sequences. Bioinformatics 1999, 15, 563-577
- Schug, J., & Overton, C.G. TESS, Transcription Element Search Software on WWW. Technical Report CBIL-TR-1997-1001-v0.0, of the Computational Biology and Informatics Laboratory, School of Medicine, University of Pennsylvania, 1997
- Neuwald, A.F., Liu, J.S., & Lawrence, C.E. Gibbs motif sampling: detection of bacterial outer membrane protein repeats. Protein Science 1995, 4: 1618-1632.
Overview
Content Tools