Page History
...
| Numbered figure captions | ||||
|---|---|---|---|---|
| ||||
|
RPKM Scaling
Standard output of mapping performed by the quantification step includes raw read counts and scaled read counts for every gene and transcript for each sample. The scaling method currently applied is reads per kilo-base of exon model per million mapped reads (RPKM) (Mortazavi et al. Nat Methods 2008). It scales the abundance estimates using exon length and millions of mapped reads and is calculated according to the formula below.

For example, suppose a transcript has 50 reads that map to it, with an exon transcript length of 7000 bp ( = 7.0 kbp), and there are 5,000,000 reads in that sample. The RPKM value is:
500/(7.000) × (1,000,000/5,000,000) = 14.28 RPKM reads
The exon length used in the formula is the sum of constituent exons, not the distance from the transcription start to the transcription stop. Moreover, as stated above, PGS uses the total number of mapped reads (not alignments!) for the RPKM calculation. In the other words, a paired-end read is counted as one read.
When adding up all of the RPKM reads for all the transcripts in a gene, they do not necessarily equal the gene level RPKM value. Only reads mapping to a transcript as exonic reads (as defined previously) will be counted as reads for that transcript. Reads partially overlap an exon or in the intronic regions are counted for gene level summarization .
During the quantification step, all aligned reads presented in the BAM file are used; no matter they are uniquely aligned or multiply aligned. If a read is uniquely aligned, then it contributes 1 count to the respective transcript. On the other hand, if a read aligns to multiple locations, then the algorithm will divide that read proportionally amongst the transcripts, depending on how likely the read maps to one or the other.
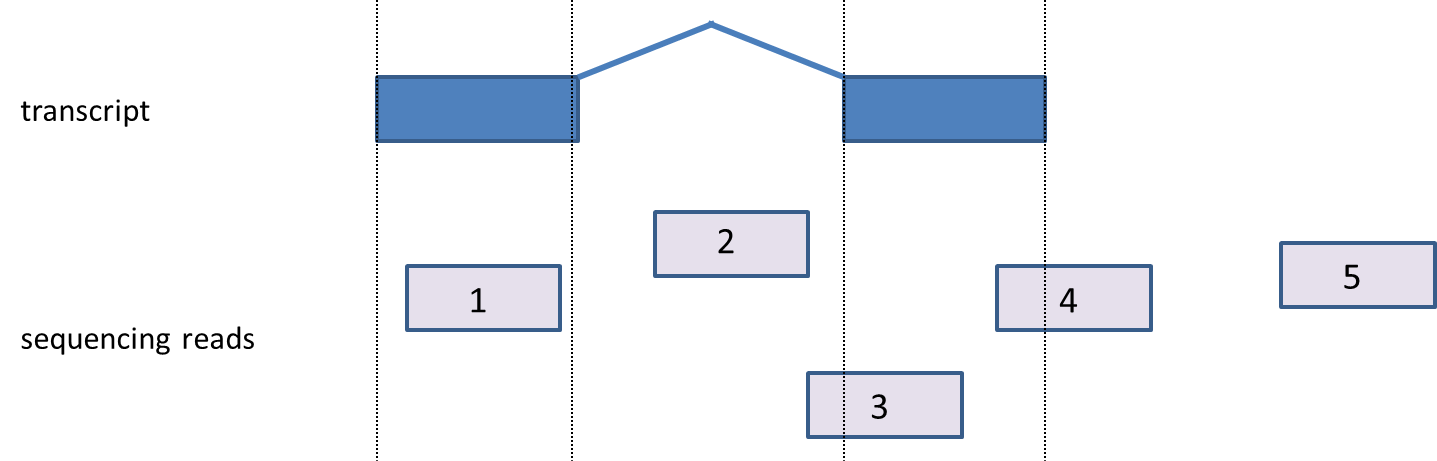
Read Compatibility
A read will be assigned to a transcript only when it is compatible. A compatible read is a read that fits the transcript model from the chosen annotation database. Compatible reads must be an exonic read (fully mapped to exon); however not all the exonic reads are compatible with a transcript, e.g., in paired end reads, both end reads have to be exonic as well as they both have to map to the same transcript; if they mapped to two different transcript or different chromosome, they are not compatible with a transcript.
...
Overview
Content Tools